


Resource-efficient pooled sequencing expands translational impact in solid tumors
Renzo G. DiNatalea,b,cRoy Manoc, Vladimir Makarovb, Nicole Ruska, Esther Drilld, Andrew Winere, Alexander Sankinf, Angela Yooc,e, Benjamin A. Freemanc, James J. Hsiehg, Ying-Bei Chenh, Jonathan A. Colemanc, Michael Bergeri, Irina Ostrovnayad, Timothy A. Chanb, Paul Russoc, Ed Reznik*a,i, A. Ari Hakimi*b,ca Computational Oncology Service, Epidemiology & Biostatistics Department, Memorial Sloan Kettering Cancer Center, New York, USA.
b Immunogenomics and Precision Oncology Platform, Memorial Sloan Kettering Cancer Center, New York, USA.
c Urology Service, Department of Surgery, Memorial Sloan Kettering Cancer Center, New York, USA.
d Biostatistics, Epidemiology & Biostatistics Department, Memorial Sloan Kettering Cancer Center, New York, USA.
e Department of Urology, SUNY Downstate Health Sciences University, Brooklyn, NY
f Department of Urology, Montefiore Medical Center and Albert Einstein College of Medicine, Bronx, NY
g Department of Medicine, University of Washington, Washington D.C., USA
h Department of Pathology, Memorial Sloan Kettering Cancer Center, New York, USA.
i Center for Molecular Oncology, Memorial Sloan Kettering Cancer Center, New York, USA.
Correspondence to: reznike@mskcc.org (ER) and hakimia@mskcc.org (AAH).
ABSTRACT
Intratumoral genetic heterogeneity (ITH) poses a significant challenge to utilizing sequencing for decision making in the management of cancer. Although sequencing of multiple tumor regions can address the pitfalls of ITH, it does so at a significant increase in cost and resource utilization. We propose a pooled multiregional sequencing strategy, whereby DNA aliquots from multiple tumor regions are mixed prior to sequencing, as a cost-effective strategy to boost translational value by addressing ITH while preserving valuable residual tissue for secondary analysis. Focusing on kidney cancer, we demonstrate that DNA pooling from as few as two regions significantly increases mutation detection while reducing clonality misattribution. This leads to an increased fraction of patients identified with therapeutically actionable mutations, improved patient risk stratification, and improved inference of evolutionary trajectories with an accuracy comparable to bona fide multiregional sequencing. The same approach applied to non-small-cell lung cancer data substantially improves tumor mutational burden (TMB) detection. Our findings demonstrate that pooled DNA sequencing strategies are a cost-effective alternative to address intrinsic genetic heterogeneity in clinical settings.
Clear cell renal cell carcinoma (ccRCC), the most common and aggressive form of kidney cancer, is characterized by extensive intratumoral heterogeneity (ITH) whereby driver mutations frequently arise only in a subset of tumor cells [1–3]. As a result of ITH, clinically informative but subclonal mutations are commonly missed by the standard practice (at our institution [4] and others [5]) of sequencing single tumor regions. In a landmark multiregional sequencing study of 101 ccRCC tumors, the TRACERx consortium reported that fifty-six percent of all detected mutations were subclonal [6], and ~20% of subclonal mutations had demonstrable clinical value either for prognostication in clinical risk models (TP53, BAP1, and PBRM1) [7], or as criteria for administration of targeted therapy (MTOR, TSC1, and PTEN) [8] (Figure 1a). Single region sequencing places a hard constraint on the sensitivity to detect and study mutations for two reasons: somatic mutation dropout (i.e. absence of a mutation in the particular tumor region sampled) and erroneous clonality assertions (i.e. attributing mutations as clonal when in fact they are only subclonal, or vice versa). Multiregional sequencing strategies address ITH by sequencing the genomic material of several spatially-separated regions of the same tumor[9]. However, due to the added sequencing expenses, this approach becomes prohibitively costly as the number of regions increases, limiting its use in practice.
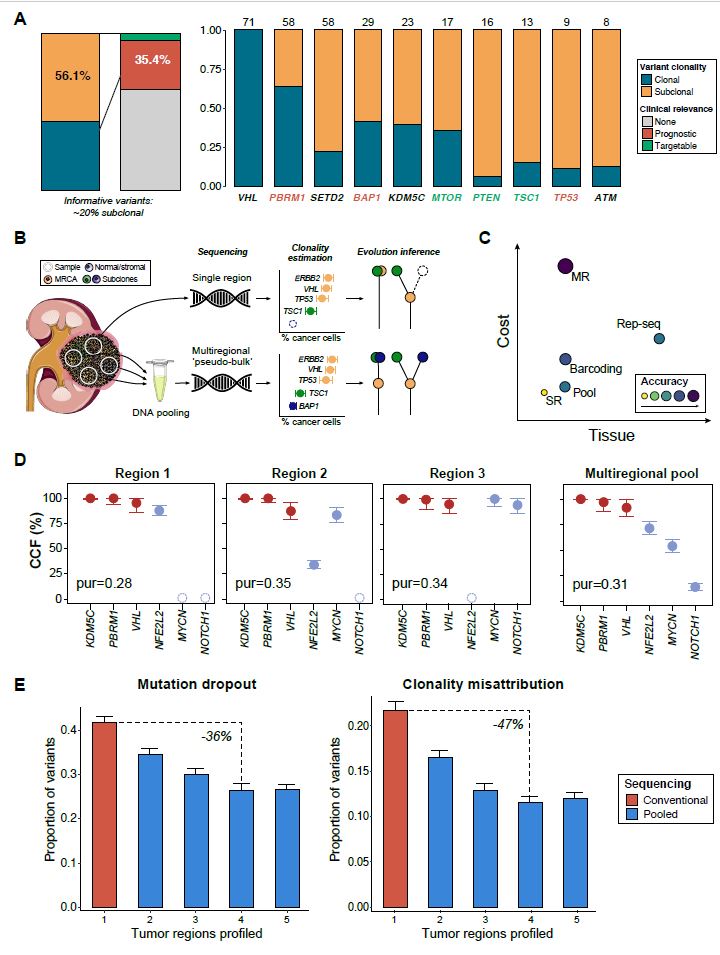
We reasoned that a more cost-effective approach to quantitatively managing ITH would be to pool samples from many regions together into a single “pseudo-bulk” before library construction (Figure 1b). Doing so would potentially ameliorate mutation dropout by increasing the likelihood of capturing a subclonal mutation, while reducing the misattribution of clonal status to mutations present only in single regions of the tumor. The benefits of a pooled approach would come at several costs: first, from diluting the sequencing bandwidth devoted to each region, and second, from loss of spatial information that would be obtained from bona fide multiregional sequencing. However, complete loss of spatial information could potentially be avoided (with an increase in cost and overhead) by barcoding DNA libraries before sequencing, an approach that has previously been demonstrated by several investigators[10]. Furthermore, pooling of tumor regions preserves precious tumor tissue which could be used for further molecular, immunohistochemical, or other profiling, and therefore is a material-efficient alternative to fully unbiased representative sequencing [11]. Direct pooling of DNA samples thus represents a flexible, cost-efficient middle-ground strategy that can be readily implemented into current pipelines without requiring additional expertise or reagents (Figure 1c).
We examined the feasibility of pooled sequencing using a deep, targeted clinical sequencing platform. For each of six ccRCC tumors, six spatially discrete regions were selected and pooled into a single sample. In parallel, we sequenced separate aliquots of the same tumor regions to standard depth, generating a ground-truth set of variant calls (Figure 1d and Supplementary figure 1a ). One case (RCC006) had no variants identified in any region and was not included in the mutational analysis ( Supplementary table 1 , Supplementary table 2). Multiregional pooled sequencing of six regions at an average depth of ~900x (150x/region) resulted in a mutation dropout rate of 4.3% (1/23 variants) and a clonality error rate of 4.5% (1/22 variants) ( Supplementary figure 2a ). Compared to single region profiling, pooled multi-regional sequencing showed a 12% lower dropout rate (95% CI: 2.0 - 22.4%, Welch t-test, p=0.02) and a 13% lower clonality error rate (95% CI: 1.2 - 24.9%, Welch t-test, p=0.03) ( Supplementary figure 2b ). Reduction in clonality misattribution was robust with the chosen cancer-cell fraction (CCF) threshold, with an estimated Matthew’s correlation coefficient (MCC) of 0.73 (with +1 indicating perfect prediction and -1 complete disagreement) ( Supplementary figure 2c, d ). Notably, all the mutations missed/misclassified were present in the tumor with the highest regional variability in purity (RCC004, with a variance 5-fold higher than the average, σ2= 0.05 vs 0.01), and none of the pooled samples had purity estimates below our quality threshold (compared to 14%, or 5/36, of the regions profiled separately). These findings demonstrate an additional potential advantage of pooled sequencing, i.e. the possibility to reduce sample failure rates during clinical sequencing.
To validate our findings and further assess the utility of this sequencing strategy, we analyzed multiregional sequencing data from the TRACERx consortium. The validation cohorts consisted of 101 individuals with a ccRCC diagnosis (median, 8 tumor regions [range, 2-75]) profiled with a sequencing panel targeting 110 cancer genes at a median depth of 612x (range, 105–1,520x) (TRACERx RCC cohort, [6]), and 100 patients with non-small cell lung cancer (TRACERx NSCLC cohort), profiled with exome sequencing at a median depth of 431x (range 83-986x) for tumor regions and 415x (range 107-765x) for the matched germline (median, 3 tumor regions, range 2-8) [12] ( Supplementary table 3 ). From our own data, we confirmed that tumor purity estimates in DNA pools were predictable in silico to high accuracy using tumor purity from single regions ( Supplementary figure 2e) Next, we simulated pooled sequencing in the TRACERx data (at equivalent depth to single-region sequencing) using a bootstrapping procedure (see Methods). Outcomes were then calculated on each random sample and averaged to produce region-number-specific estimates.
Pooled sequencing substantially decreased mutation dropout relative to single region profiling, even with the addition of just a single region (17% decrease in dropout with a pool of two regions). Similarly, we observed that this approach significantly improved our ability to correctly assign clonality to observed mutations, with a 24% drop in clonality assignment error with the addition of a single region to a pool (Figure 1e). When evaluating these same outcomes at the patient-level (i.e. proportion of individuals with at least one variant dropped/misclassified), we observed that pooled sequencing with a single additional region would result in a 14% decrease in both the number of patients subject to mutation dropout and the number affected by misattribution of clonality ( Supplementary figure 3b, d). Consistent with the rarity of spatially-delimited low-allele-frequency mutations in ccRCC (arising in cancer genes), we observed a negligible number of false-negative mutation calls with a higher number of regions (Figure 2a). No differences were observed between tumor pools of four regions and those with higher numbers when evaluating their reliability when attributing mutation clonality, however, this result was found specific to the tumor type context ( Supplementary figure 4a, b).
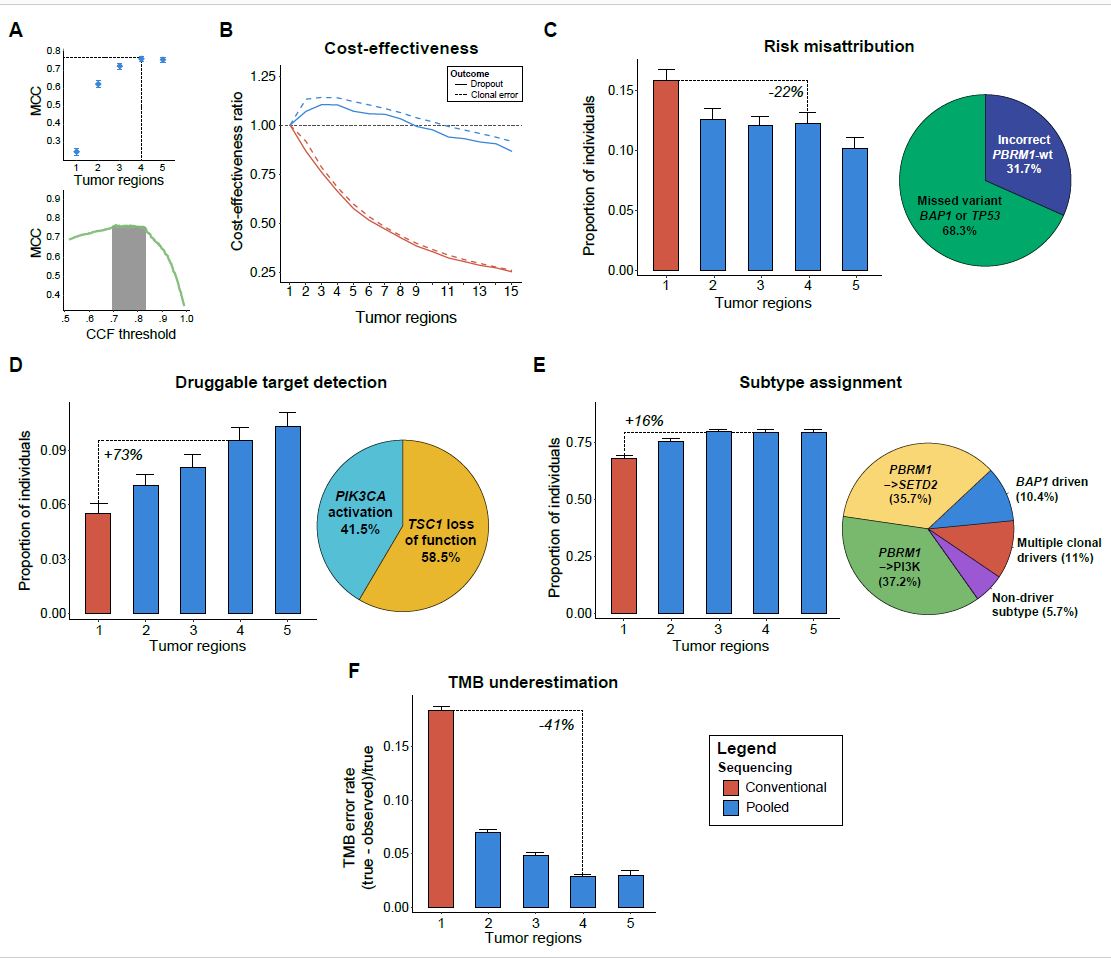
Importantly, because the financial cost of next-generation sequencing (NGS) assays is dominated by sequencing costs (i.e. related to library size due to depth and breadth) rather than sample processing and genomic material extraction, obtaining DNA from multiple regions and mixing them into a single pseudo-bulk would result in minimal additions to the total cost ( Supplementary Figure 5a ). Given that our direct pooling approach requires no additional reagents (nor modifications to the computational infrastructure), it occupies a flexible middle-ground compared to bona fide multiregional sequencing and full-scale mixing of left-over tumor tissue [10,11]. We defined a metric of cost-effectiveness as the change in mutation dropout (or clonality) per tumor relative to the change in cost ( Supplementary figure 5 b, c ). Using cost estimates for targeted panel sequencing from our own institution, we compared the cost-effectiveness of conventional and pooled multi-regional sequencing relative to single-region profiling. Pooled sequencing (of 2 to 4 tumor regions) was found to be ~10% more cost-effective than single-region profiling both for mutation detection and clonality assessment, while the opposite was observed with conventional multi-regional sequencing. Notably, the added benefit of pooled sequencing was lost when pooling 10 regions or more (Figure 2b).
We next examined the translational utility of pooling discrete regions of a tumor in the management of ccRCC by several metrics. In patients with metastatic ccRCC receiving first-line treatment with tyrosine kinase inhibitors, the mutation status (irrespective of clonal status) of PBRM1, BAP1, and TP53 is of prognostic significance [7], and dropout of somatic variants in these genes therefore affects risk stratification. We observed that risk stratification would be affected in 10% of the TRACERx RCC patients if only a single region were sequenced. Pooled sequencing of 4 regions corrected the risk stratification in 4% of patients, effectively reducing the baseline error in risk stratification by 22% (Figure 2c). Furthermore, the presence of mutations in a subset of genes represents potential therapeutically actionable targets and/or eligibility criteria for clinical trials. Pooled sequencing (of 4 regions) significantly increased the number of patients identified with such mutations by more than 70% (from 6% to 10%) (Figure 2d).
Independent of its translational value, pooled sequencing provides a cost-effective lens onto patterns of ITH. Recent work by the TRACERx consortium in ccRCC has proposed “evolutionary subtypes” based on the presence and clonality of mutations in five genes (VHL, PBRM1, SETD2, BAP1 and PTEN). We examined our capacity to correctly assign evolutionary subtypes in pooled sequencing according to the heuristics outlined by Turajlic and colleagues [6]. Pooling four tumor regions increased the correct evolutionary subtype assignment by 16%, with the majority of missed subtypes corresponding to the ‘PBRM1→PI3K’ and ‘PBRM1→SETD2’subtypes, with relatively good outcomes (Figure 2e). Pooled sequencing thus represents a potential strategy for the interrogation of subclonal mutational diversity and inference of evolutionary trajectories, which have further implications for patient outcomes.
Finally, we explored the translational value of pooled sequencing in the context of an entirely different disease and sequencing platform. In a cohort of 100 NSCLC patients from the TRACERx consortium, we evaluated in silico the utility of pooled sequencing in accurately quantifying tumor mutation burden (TMB); this measure is employed as a biomarker for response to immunotherapy in this disease. While single region sequencing underestimates total tumor mutation burden by nearly 20%, the addition of a single region to a DNA pool reduced this effect by 41% (Figure 2f). Since the clonality of neoantigens is an emerging determinant of T cell immunoreactivity [13], and given that TMB and neoantigen load are highly correlated [14], accurate assessment of mutation burden with multiregional approaches may improve prognostication in the context of immunotherapy for NSCLC. However, the ability of a clonality-aware TMB measure to predict response to immune-checkpoint blockade will need to be evaluated in this context, as it is currently optimized to the single region setting[15].
Intratumoral heterogeneity is a fundamental hurdle in the genomically-informed delivery of care to cancer patients. In ccRCC, such heterogeneity is so pervasive that it confounds the accurate identification of the small set of driver mutations of therapeutic relevance. Our proposed approach of direct pooled DNA sequencing from several tumor regions overcomes some of these issues at a fraction of the cost of bona fide multiregional profiling; and it does so without excess use of precious tissue material, preserving it for subsequent profiling studies. Pooling thus represents a viable and cost-effective strategy to overcome ITH during clinical sequencing. Importantly, the overhead costs for both single region and pooled sequencing, including sample acquisition, data handling, storage, and analysis, are largely the same, as the size of the sequencing library remains identical. Furthermore, multiregional DNA pooling allows for the inclusion of additional processing steps before sequencing, providing an extra degree of flexibility when adjusting this approach to different clinical scenarios. Finally, by mixing regions of variable purity, we also envision that pooled sequencing may ameliorate the ~3% of tumor samples (~300/10,000 per year total) which currently fail clinical sequencing at our institution due to excessively low tumor purity, thus increasing resource utilization efficiency [16].
Our current analysis is limited to single nucleotide variants and indels. However, copy number variants (CNVs) could be similarly evaluable by pooled sequencing. However, the relatively low density of heterozygous SNPs tiling the genome in targeted sequencing platforms renders the attribution of clonality to CNVs extremely challenging[9]. One might speculate that ongoing refinement of targeted sequencing panels, or the use of broader panels could create new opportunities for copy-number analysis from pooled sequencing.
Although sequencing technologies have greatly expanded our knowledge of the molecular mechanisms behind the development and progression of RCC, these discoveries have yet to be translated into tangible clinical benefits during treatment selection or prediction of therapy response. However, it is unclear if this lack of clinical applicability is indeed inherent to the biology of the disease or a result of pervasive sampling biases in past genomic studies that have profiled a single tumor region. Therefore, it is imperative to expand on these initiatives and consider novel sequencing strategies that allow for multi-region assessment of heterogeneous tumors.
SUPPLEMENTARY MATERIALS
- SUPPLEMENTARY FIGURE LEGENDS
-
DATA AVAILABILITY
All the processed data and statistical code needed to reproduce the findings in this study have been provided in the supplementary materials as well as in a publicly-available repository ( https://github.com/reznik-lab/DNApooling_RD ). The raw sequencing data (MSK-IMPACT) produced in this study are deposited on the Sequence Read Archive (SRA) under the accession number PRJNA633220. Data from the validation sets are available in the supplementary materials of the original TRACERx publications [6,12], only the filtered/annotated versions are provided with this manuscript. Any other relevant data is available from the corresponding authors upon reasonable request.
ACKNOWLEDGMENTS
We thank Charles Swanton, Samra Turajlic, Nicholas McGranahan, Kevin Litchfield and all the members of the TRACERx consortium for facilitating the mutation data used in the study. We thank the Reznik and Chan Lab members for their helpful discussions. We thank the staff and physicians of the MSK Department of Medicine Kidney Program and the Urology Service for their helpful suggestions. We acknowledge the use of the Integrated Genomics Operation Core, funded by the NIH/NCI Cancer Center Support Grant (CCSG, P30 CA008748), Cycle for Survival, and the Marie-Josée and Henry R. Kravis Center for Molecular Oncology. This work was in part supported by grants NIH R35 CA232097 and DOD KC180165 (TAC), as well as the DOD Kidney Cancer Research Program W81XWH-18-1-0318 and the Kidney Cancer Association Young Investigator Award. (ER).This work was also supported by the Mellnikoff Fund (TAC) and the Weiss Family Fund (AAH).
COMPETING INTERESTS
T.A.C. is a co-founder of Gritstone Oncology and holds equity. T.A.C. holds equity in An2H. T.A.C. acknowledges grant funding from Bristol-Myers Squibb, AstraZeneca, Illumina, Pfizer, An2H, and Eisai. T.A.C. has served as an advisor for Bristol-Myers, MedImmune, Squibb, Illumina, Eisai, AstraZeneca, and An2H. He also holds ownership of intellectual property on using tumor mutation burden to predict immunotherapy response, with pending patent, which has been licensed to PGDx.
The rest of the authors have no conflicts to disclose.
AUTHOR CONTRIBUTIONS
Sample and patient data procurement: AW, AS, YC, PR, JC, AY, BAF
Data processing: VM, RGD, MB, TAC
Statistical analysis: RGD, ED, IO, ER
Manuscript preparation: RGD, RM, NR, ER, AAH
Oversight: AAH, ER, TAC, PR, JH
KEYWORDS: • cancer genomics • cancer evolution • intratumoral heterogeneity • next-generation sequencing • somatic mutation, clonality •
REFERENCES
- 1. Hsieh JJ, Purdue MP, Signoretti S, Swanton C, Albiges L, Schmidinger M, et al. Renal cell carcinoma. Nat Rev Dis Primers. 2017;3:17009.
- 2. Ricketts CJ, Marston Linehan W. Intratumoral heterogeneity in kidney cancer [Internet]. Nature Genetics. 2014. p. 214–5. Available from: http://dx.doi.org/10.1038/ng.2904
- 3. McGranahan N, Swanton C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell. 2017;168:613–28.
- 4. Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J Mol Diagn. 2015;17:251–64.
- 5. AACR Project GENIE Consortium. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017;7:818–31.
- 6. Turajlic S, Xu H, Litchfield K, Rowan A, Horswell S, Chambers T, et al. Deterministic Evolutionary Trajectories Influence Primary Tumor Growth: TRACERx Renal. Cell. 2018;173:595–610.e11.
- 7. Voss MH, Cheng Y, Marker M, Kuo F, Choueiri TK, Hsieh JJ, et al. Incorporation of PBRM1, BAP1, TP53 mutation status into the Memorial Sloan Kettering Cancer Center (MSKCC) risk model: A genomically annotated tool to improve stratification of patients (pts) with advanced renal cell carcinoma (RCC) [Internet]. Journal of Clinical Oncology. 2018. p. 639–639. Available from: http://dx.doi.org/10.1200/jco.2018.36.6_suppl.639
- 8. Chakravarty D, Gao J, Phillips SM, Kundra R, Zhang H, Wang J, et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis Oncol [Internet]. 2017;2017. Available from: http://dx.doi.org/10.1200/PO.17.00011
- 9. Turajlic S, Sottoriva A, Graham T, Swanton C. Resolving genetic heterogeneity in cancer. Nat Rev Genet. 2019;20:404–16.
- 10. Zhang X, Garnerone S, Simonetti M, Harbers L, Nicoś M, Mirzazadeh R, et al. CUTseq is a versatile method for preparing multiplexed DNA sequencing libraries from low-input samples. Nat Commun. 2019;10:4732.
- 11. Litchfield DK, Stanislaw S, Spain L, Gallegos L, Rowan A, Schnidrig D, et al. Representative Sequencing: Unbiased Sampling of Solid Tumor Tissue [Internet]. SSRN Electronic Journal. Available from: http://dx.doi.org/10.2139/ssrn.3404257
- 12. Jamal-Hanjani M, Wilson GA, McGranahan N, Birkbak NJ, Watkins TBK, Veeriah S, et al. Tracking the Evolution of Non-Small-Cell Lung Cancer. N Engl J Med. 2017;376:2109–21.
- 13. McGranahan N, Furness AJS, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351:1463–9.
- 14. Chan TA, Yarchoan M, Jaffee E, Swanton C, Quezada SA, Stenzinger A, et al. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann Oncol. 2019;30:44–56.
- 15. Budczies J, Allgäuer M, Litchfield K, Rempel E, Christopoulos P, Kazdal D, et al. Optimizing panel-based tumor mutational burden (TMB) measurement. Ann Oncol. 2019;30:1496–506.
- 16. Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23:703–13.
- 17. DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–8.
- 18. Kim S, Scheffler K, Halpern AL, Bekritsky MA, Noh E, Källberg M, et al. Strelka2: Fast and accurate variant calling for clinical sequencing applications [Internet]. Available from: http://dx.doi.org/10.1101/192872
- 19. Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–76.
- 20. Rimmer A, Phan H, Mathieson I, Iqbal Z, Twigg SRF, WGS500 Consortium, et al. Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat Genet. 2014;46:912–8.
- 21. Sherry ST. dbSNP: the NCBI database of genetic variation [Internet]. Nucleic Acids Research. 2001. p. 308–11. Available from: http://dx.doi.org/10.1093/nar/29.1.308
- 22. Consortium T 1000 GP, The 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing [Internet]. Nature. 2010. p. 1061–73. Available from: http://dx.doi.org/10.1038/nature09534
- 23. Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration [Internet]. Briefings in Bioinformatics. 2013. p. 178–92. Available from: http://dx.doi.org/10.1093/bib/bbs017
- 24. Barnell EK, Ronning P, Campbell KM, Krysiak K, Ainscough BJ, Sheta LM, et al. Standard operating procedure for somatic variant refinement of sequencing data with paired tumor and normal samples. Genet Med. 2019;21:972–81.
- 25. Shen R, Seshan VE. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 2016;44:e131.
- 26. Bielski CM, Zehir A, Penson AV, Donoghue MTA, Chatila W, Armenia J, et al. Genome doubling shapes the evolution and prognosis of advanced cancers. Nat Genet. 2018;50:1189–95.
- 27. Jiang Y, Qiu Y, Minn AJ, Zhang NR. Assessing intratumor heterogeneity and tracking longitudinal and spatial clonal evolutionary history by next-generation sequencing. Proc Natl Acad Sci U S A. 2016;113:E5528–37.
- 28. Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30:413–21.
- 29. McGranahan N, Favero F, de Bruin EC, Birkbak NJ, Szallasi Z, Swanton C. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci Transl Med. 2015;7:283ra54.
- 30. Turajlic S, Xu H, Litchfield K, Rowan A, Chambers T, Lopez JI, et al. Tracking Cancer Evolution Reveals Constrained Routes to Metastases: TRACERx Renal. Cell. 2018;173:581–94.e12.
- 31. Chicco D, Jurman G. The advantages of the Matthews correlation coefficient (MCC) over F1 score and accuracy in binary classification evaluation. BMC Genomics. 2020;21:6.
- 32. Samstein RM, Lee C-H, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet. 2019;51:202–6.

