Submitted - October 18, 2022 | Revised December 4, 2022 Accepted - December 7, 2022 | | ePublished - December 31, 2022
https://doi.org/10.52733/KCJ21n4-or1

ABSTRACT
Fumarate hydratase-deficient renal cell carcinomas are an aggressive form of kidney cancer that often results in poor prognosis and high fatality rates. The implications of somatic mutations are not well described, and standard treatment has not been established for this renal cell carcinoma subtype. Further molecular characterization of fumarate hydratase-deficient renal cell carcinomas could potentially help to identify biomarkers that can be exploited with future targeted therapies. 2199 renal cell carcinomas were analyzed by DNA sequencing (592-gene panel) and whole- transcriptome sequencing and 40 tumors were identified with pathogenic FH mutations. Co-occurrence of mutation with other cancer-related genes were assessed along with immune profiles and immunotherapy biomarkers. Fumarate hydratase-deficient renal cell carcinomas had a lower prevalence of comutation with common renal cell carcinoma driver mutations such as VHL and chromatin remodeling genes when compared to wild type renal cell carcinoma. Conversely, prevalence of several cancer-related genes (MAX, BRCA1, PMS2, BRAF, NF2, and AKT1) was higher in fumarate hydratase-deficient renal cell carcinomas. Immunotherapy biomarkers (mismatch repair deficiency and tumor mutational burden) were detected at low frequency in mutant and wild type renal cell carcinomas, while PDL1 expression occurred at higher frequency in fumarate hydratase-deficient renal cell carcinomas. Fumarate hydratase-mutated kidney tumors may have a different mutational and immune landscape than wild type tumors. The absence of VHL mutations in a significant number of fumarate hydratasedeficient renal cell carcinomas suggest that FH mutations may drive tumorigenesis using distinct angiogenic pathways. Our study highlights potential therapeutic implications that will require further study.
INTRODUCTION
Fumarate hydratase (FH) is a
key component of the Krebs
cycle, and loss of FH function
leads to multiple disorders, including
aggressive forms of cancer.
Heterozygous germline mutations
leading to FH deficiency are associated
with hereditary predisposition to
multiple tumors.1,2 Fumarate can act
as an oncometabolite by inhibiting
multiple α-ketoglutarate (α-KG)-dependent
dioxygenases, which in turn
stabilizes hypoxia inducible factor 1
subunit alpha (HIF1α), creating a
state of pseudohypoxia that leads to
angiogenesis and tumor growth.3-5
Thus, functional FH is often referred
to as a tumor suppressor. FH-deficient
renal cell carcinoma (RCC) is
an aggressive form of renal cancer
that was first described as part of hereditary
leiomyomatosis and renal
cell cancer (HLRCC) syndrome.1,6
Although it was initially regarded
as papillary type II RCC, the term
FH-deficient RCC is now preferred
as it can present in other histological
subtypes of RCC. Germline mutations
in the FH gene impart a high
risk for developing tumors at an early
age,6-8 and HLRCC patients who
develop RCC have significantly shorter survival when diagnosed with
advanced stage compared to early
stage.9 To improve early detection
rates and survival, regular screening
for RCC in HLRCC individuals
is recommended. 10,11 FH-deficient
RCC can also develop by sporadic
loss-of-function mutation in the FH
gene; however, there is no consensus
on the implications of FH alterations
in RCC outside of the HLRCC
syndrome.12,13
F H-deficient t umors o ften
result in poor prognosis and high
fatality rates, while standard
treatment in the advanced
disease stage setting has not been
established for this aggressive RCC
subtype.9 While immunotherapy
combination strategies have
improved outcomes for patients
with clear cell RCC (ccRCC), 14 they
have not been heavily tested in
variant histologies. It is also unclear
how the mutational landscape in
FH-deficient patients augments
sensitivity to immunotherapy or
other targeted therapies. Therefore,
further characterization of FHdeficient
RCC with comprehensive
molecular profiling could potentially
help to identify biomarkers that can
be exploited with future targeted
therapies. The goal of this study is
to enhance our knowledge of the
molecular landscape of FH-deficient
renal tumors (hereafter referred to
as FH-mut tumors) in relation to
wild type tumors (WT) lacking FH
alterations. From DNA and RNA
analysis, co-occurrence of mutation
with other cancer-related genes were
assessed along with immune profiles and immunotherapy biomarkers.
MATERIALS AND METHODS
Sample collection from participants A total of 2199 RCCs underwent comprehensive tumor profiling at Caris Life Sciences (Phoenix, AZ, USA). This study was conducted in accordance with guidelines of the Declaration of Helsinki, Belmont Report, and U.S. Common Rule. In keeping with 45 CFR 46.101 (b), this study was performed utilizing retrospective, deidentified clinical data from patients with renal cancer. Therefore, this study was considered Institutional Review Board exempt and no patient consent was necessary from the subjects.Next-Generation Sequencing (NGS)
NGS was performed on genomic DNA isolated from formalin-fixed paraffin-embedded (FFPE) tumor samples using the NextSeq or NovaSeq platform (Illumina, Inc., San Diego, CA, USA). For NextSeq, a custom-designed SureSelect XT assay was used to enrich 592 wholegene targets (Agilent Technologies, Santa Clara, CA). All variants were detected with >99% confidence based on allele frequency and amplicon coverage, with an average sequencing depth of coverage of >500x and an analytic sensitivity of 5%. For NovaSeq, a hybrid pull-down panel of baits designed to enrich for more than 700 clinically relevant genes at high coverage (>500x) and high read-depth was used, along with another panel designed to enrich for an additional >20,000 genes at lower depth (>250x). Genetic variants identified were interpreted by board-certified molecular geneticists and categorized as pathogenic, likely pathogenic, or variant of unknown significance, according to ACMG standards. All variants were detected with greater than 99% confidence based on allele frequency and amplicon coverage, with an average sequencing depth of coverage of greater than 500 and an analytic sensitivity of 5%. For RNA sequencing (RNASeq), biotinylated RNA baits were hybridized to the synthesized and purified cDNA targets and the baittarget complexes were amplified in a post capture PCR reaction. The resultant libraries were quantified, normalized, and the pooled libraries were denatured, diluted, and sequenced; the reference genome used was GRCh37/hg19 and analytical validation of this test demonstrated ≥97% positive percent agreement (PPA), ≥99% negative percent agreement (NPA) and ≥99% overall percent agreement (OPA) with a validated comparator method. Transcripts per million (TPM) values were generated using the Salmon expression pipeline for transcription counting. Multiple test platforms were used to determine the microsatellite instability (MSI) or mismatch repair (MMR) status of the tumors, including fragment analysis (FA), IHC, and NGS. For IHC the following antibodies were used: M1 antibody for MLH1 (Roche Diagnostics, Belmont, CA, USA), G219-1129 antibody for MSH2 (Roche Diagnostics, Belmont, CA, USA), 44 antibody for MSH6 (Thermo Fisher Scientific, Carlsbad, CA, USA), and EPR3947 antibody for PMS2 (Abcam, Waltham, MA, USA). For NGS, 7,000 target microsatellite loci were examined and compared to the reference genome hg19). The tumor was determined MSI-high (MSI-H) by FA if two or more mononucleotide out of the five markers included in the assay were abnormal; the tumor was considered mismatch repair deficient (dMMR) by IHC if complete absence of protein expression of any of the four proteins was observed; the tumor was considered MSI-H by NGS by a threshold of 46 or more altered loci per tumor. MSI or MMR status of the tumor was determined in the order of IHC, FA, and NGS. TMB was measured by counting all non-synonymous mutations found per tumor that had not been previously described as germline alterations in dbSNP151, Genome Aggregation Database (gnomAD) databases or benign variants identified by Caris geneticists. A cutoff point of >=10 mutations per MB was used based on the KEYNOTE-158 pembrolizumab trial.15,16 Immune cell fractions were calculated from deconvolution of bulk RNA-Seq data using the QuantiSeq computational pipeline.17 Interferon-gamma score (IFN score) was calculated based on weighted sum of TPM values of 18 genes as previously described.18
Immunohistochemistry (IHC)
IHC was performed on full FFPE sections of glass slides using automated staining techniques, per the manufacturer’s instructions, and were optimized and validated per CLIA/CAO and ISO requirements. The staining was scored for intensity (0 = no staining; 1+ = weak staining; 2+ = moderate staining; 3+ = strong staining) and staining percentage (0–100). Results were categorized as positive or negative by defined thresholds specific to each marker based on published clinical literature that associates biomarker status with patient responses to
therapeutic agents. A boardcertified
pathologist evaluated all
IHC results independently. The
primary antibody used against PDL1
was SP142 (Spring Biosciences,
San Francisco, CA, USA). The
staining was regarded as positive
if its intensity on the membrane
of the tumor cells was 2+ (on a
semiquantitative scale of 0–3: 0 for
no staining, 1+ for weak staining,
2+ for moderate staining, or 3+ for
strong staining) and the percentage
of positively stained cells was >5%.
Statistical Analysis
The molecular features of tumors carrying pathogenic or likely pathogenic (P/LP) and WT FH tumors were compared. Categorical data was assessed using a chisquare or Fisher Exact test, where appropriate. Immune cell abundance in the tumor micro-environment were estimated using the method described above17 and significance was tested using a nonparametric Wilcoxon rank-sum test. Gene expression for immune checkpoint genes was normalized to the median gene expression in the control group and fold change was calculated; significance was tested using nonparametric Wilcoxon ranksum test. P-values were adjusted for multiple hypothesis testing by Bonferroni or Benjamini-Hochberg. All statistical analyses were two sided at a significance level set to 0.05.
Availability of data and materials
The deidentified sequencing data are owned by Caris Life Sciences. The datasets generated during and analyzed during the current study are available from the authors upon reasonable request and with permission of Caris Life Sciences.
RESULTS Basic Cohort Description
A total of 2199 RCCs were included in this analysis, with ages ranging from two to 90 years old, and a median age of 63. In this cohort, 29.4% were females and 70.6% were males. Nine hundred forty-four (42.9%) samples were from a primary tumor, and 1240 (56.4%) samples were from a metastatic or distant site (Table 1). Among all tumors, 2143 (97.4%) of the tumors did not harbor a FH mutation (wildtype, WT), 40 (1.82%) tumors had a pathogenic mutation or likely pathogenic mutation (P+LP-mt), and 16 (0.73%) tumors had a variant of unknown significance (VUS-mt) (Table 1). There was no significant difference in gender distribution among the various FH-mutant groups (P+LP-mt vs VUS-mt vs WT: 30% vs 37.5% vs 29.4% for female and 70% vs 62.5% vs 70.6% for male). However, more P+LP-mt tumors were from the renal primary site compared to VUS-mt or WT tumors (52.5% vs 12.5% vs 43%) (Table 1). We sorted the data to differentiate two main histological subtypes of renal cancer: tumors clearly noted as clear cell histology (ccRCC) and those with unclear or other histological subtypes (non-ccRCC). Among the 704 ccRCC tumors included in this analysis, 11 (1.56%) were P+LP-mt. Among 1495 non-ccRCC tumors, 29 (1.94%) were P+LP-mt tumors.
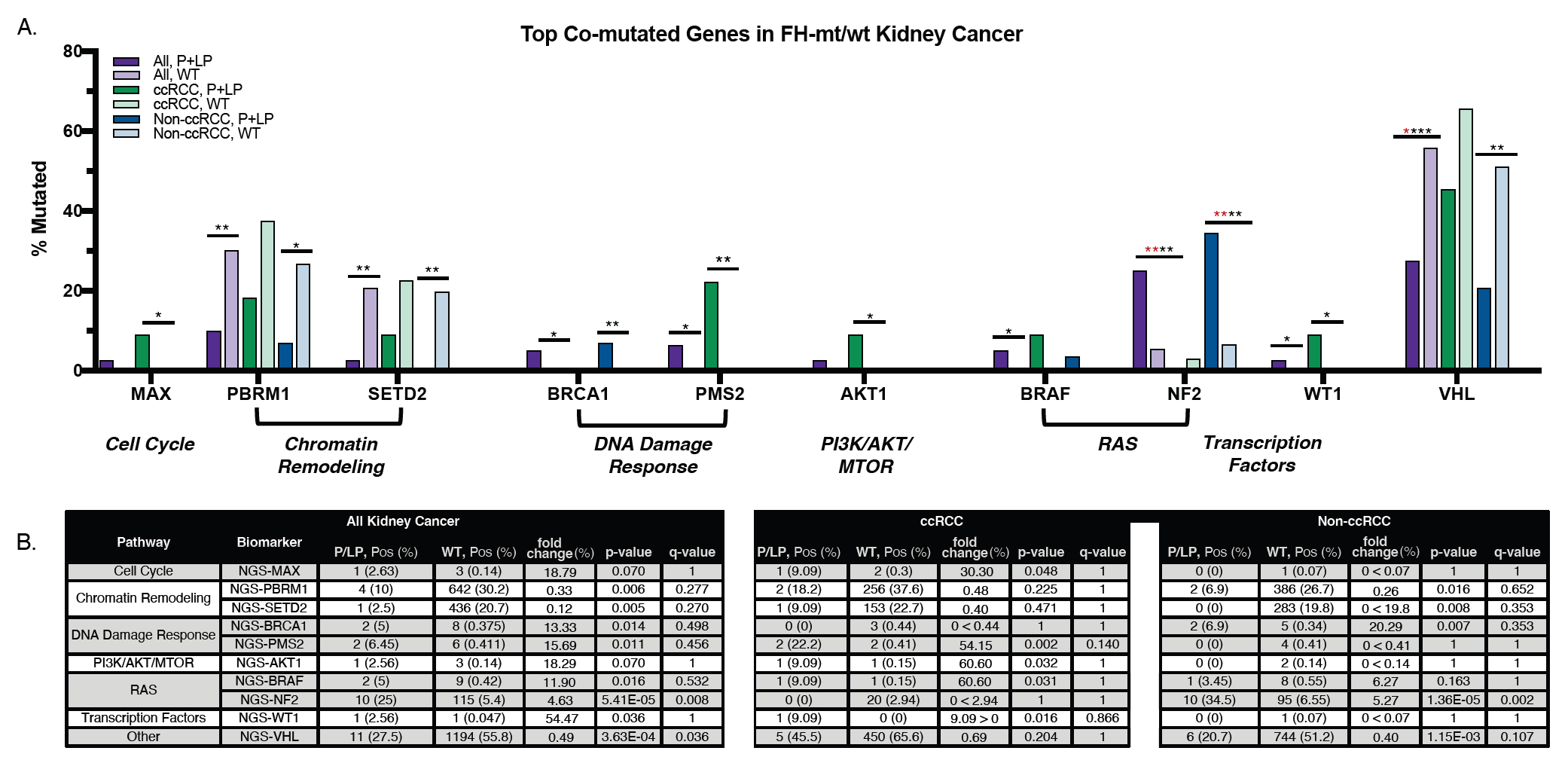
Mutational landscape of FHmutated Renal Cell Carcinoma
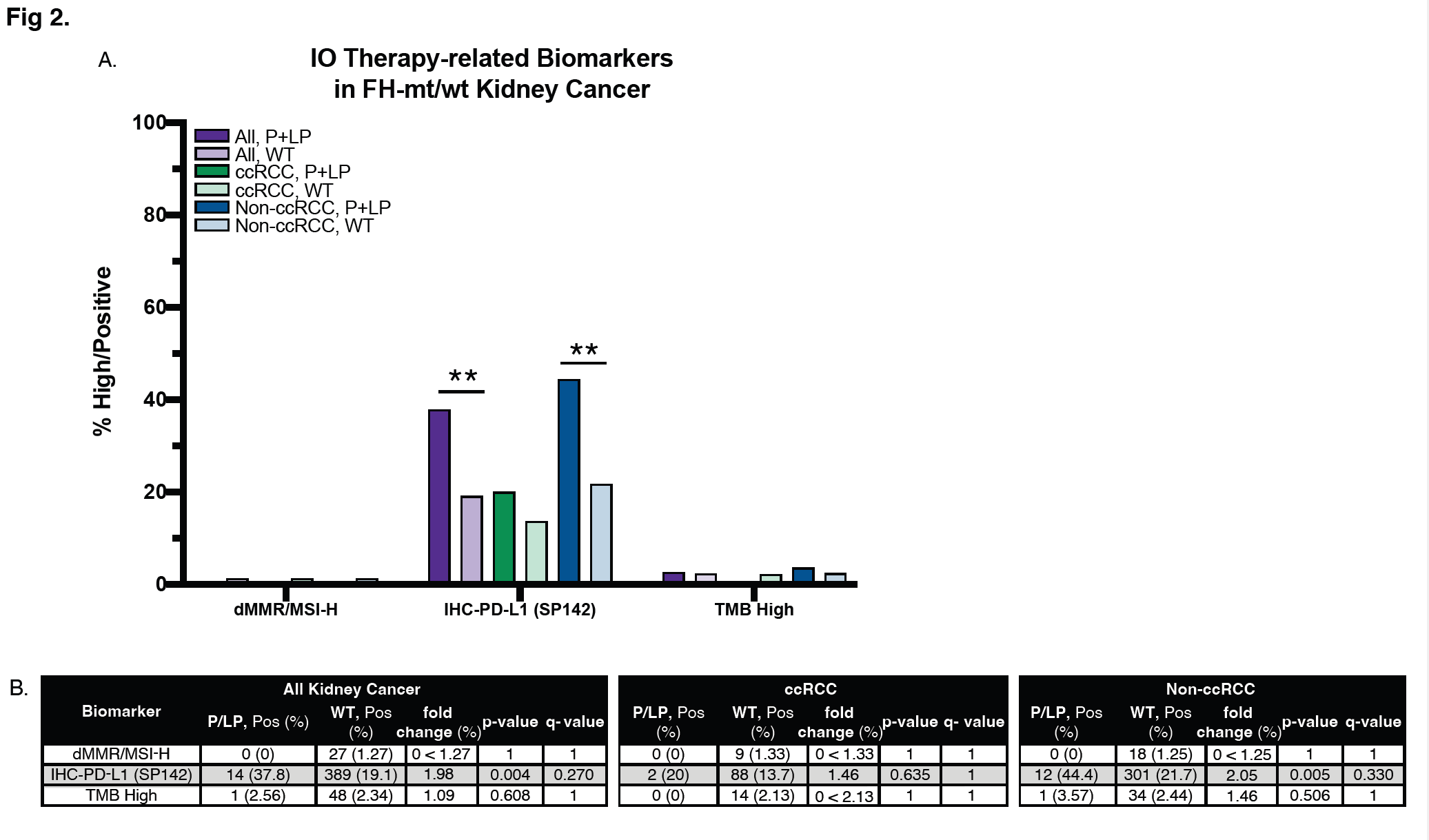
When examining the co-occurrence of cancer-related mutations in our cohort (Figure 1), a lower prevalence of FH co-mutation was observed with VHL in P+LP-mt tumors compared to WT tumors overall (27.5% vs 55.8%, p-value=3 .63E - 04). The lower frequency of VHL comutation was observed in both ccRCC to non-ccRCC, but the difference between P+LP-mt and WT tumors was more significant in the latter (p-value=1.15 E-03). A similar pattern was observed with chromatin remodeling genes, with lower prevalence of comutation with PBRM1 and SETD2 in P+LP-mt tumors (10% vs 30.2%, p-value=0.006; and 2.5% vs 20.7%, p-value=0.005, respectively). Again, this difference was observed in both ccRCC and non-CCRCC, but more significantly in the latter. (p-value=0.016 for PBRM1; p-value=0.008 for SETD2). Conversely, co-mutation prevalence in P+LP-mt tumors was higher in several cancer-related genes. With the cell cycle gene, MAX, co-mutations were detected at a higher rate in P+LP-mt tumors compared to WT tumors (2.63% vs 0.14%, p-value=0.07) and observed only in the ccRCC histology subtype (p-value=0.048). BRCA1 comutations were higher in P+LP-mt tumors (5% vs 0.38, p-value=0.014) only in the non-ccRCC histology subtype. Alternately, PMS2 comutations were higher in P+LPmt tumors (6.45% vs 0.41%, p-value=0.11) only in the ccRCC histology subtype. Among genes in the RAS pathway, higher BRAF co-mutations in P+LP-mt tumors (5% vs 0.42%, p-value=0.016) were observed in both histology subtypes, yet more significantly in ccRCC (p-value=0.031). NF2 comutations were higher in P+LP-mt tumors compared to WT (25% vs 5.4%, p-value=5.41E-05) only in non-ccRCC. Other prevalent comutations that increased in P+LP-mt tumors were detected in AKT1 (2.56% vs 0.14%, p-value=0.07) and WT1 (2.56% vs 0.05%, p-value=0.036), both of which were only observed in ccRCC (p-value=0.032 for AKT1; p-value=0.016 for WT1) (Figure 2). Distribution of mutations among ccRCC and non-ccRCC, and primary vs metastatic tumors is further illustrated in Supplemental Figure 1. Mismatch repair deficiency, or MSI-high status, was found at low frequency (1.27%) in WT tumors, and not observed in P+LP-mt tumors (Figure 2). TMB-high status was also very low in both P+LP-mt tumors and WT tumors with no significant difference between the groups (2.56% vs 2.34%, respectively. p-value=0.608). On the other hand, PD-L1 expression was detected at a higher frequency of P+LP-mt tumors compared to WT tumors (37.8% vs 19.1%, p-value=0.004). This pattern was observed in both ccRCC and nonccRCC, although only significant in the latter (p-value=0.005).
Immune landscape of FHmutated Renal Cancer
Immune infiltration analysis indicated P+LP-mt tumors had less NK cell and M2 macrophage infiltration compared to WT tumors (Figure 3). This pattern was observed in both ccRCC and nonccRCC, though significant only in non-ccRCC. Most other immune cell types demonstrated slightly lower infiltration in P+LP-mt tumors, with the exception of dendritic cells which were higher in ccRCC and lower in non-ccRCC. Most differences in expression levels of immune checkpoint genes were not statistically significant, except for lower expression of LAG3 and PDCD1 in P+LP-mt tumors relative to WT tumors. P+LP-mt tumors exhibited higher expression of CD274 in both histology subtypes. Although not statistically significant, expression of CTLA4 and IFNG were highest in ccRCC tumors (Figure 3A, 3D). The IFN signature was higher in ccRCC, but significantly lower only in nonccRCC (Figure 3C, 3D).
DISCUSSION
Numerous reports have described distinct prognostic indicators and therapeutic options for specific RCC subtypes,19-22 yet most of the available literature and clinical trials have not been conducted in the context of genomic mutations, including FH status. Although the relatively small numbers of FHmut tumors within each histology subgroup limits the conclusions that can be drawn, some of the results point to possible trends that may warrant further investigation. Examination of co-mutated genes revealed that some mutations which are commonly found in RCC (VHL, PBRM1, SETD2) often do not occur concurrently in FH-mut tumors. This suggests that loss of FH may be an important driver of tumorigenesis in RCC independently of classic RCC drivers. The presence or absence of such co-mutations may also affect the efficacy of targeted therapies that have recently been developed for RCC naively of FH status. For example, PBRM1 deficiency has been associated with clinical benefit from ICI therapy,23 and recently transcriptome-based molecular profiles integrated with PBRM1 status and angiogenesis signatures have been developed to further facilitate clinical guidance.23-25 With advances in the use of molecular subtypes for predictive value, FH status and associated mutations may therefore be considered in the cases where common RCC driver mutations are absent.
We observed VHL mutations occurring most frequently in ccRCC, which agrees with reports citing the prevalence of VHL mutations in ccRCC. In our study, VHL mutations were decreased in all FH-mut tumors with the most significant decrease in non-ccRCC (Figure 1). Neoangiogenesis linked to the von- Hippel Lindau (VHL) gene has been commonly implicated as the main pathway of tumorigenesis in renal cancers. 6 As VHL inactivation leads to HIF activation, subsequent tumorigenesis may occur in a similar fashion to what has subsequently been attributed to FH deficiency. Reduced FH expression in ccRCC was demonstrated via HIF stabilization to increase VEGF production.27 Thus, HIF inhibitors and other drugs targeting metabolic pathways have become the basis of emerging targeted therapies against RCC and may also be applicable to FH-deficient tumors.28,29 Antiangiogenic tyrosine kinase inhibitors (TKI) of the VEGF pathway led to improved outcomes in several studies involving metastatic RCCs.30-33 Further improvements have also been demonstrated when TKIs were combined with immunotherapies.34 The similar angiogenic effects that FH mutations and VHL mutations have indicates that FH-mut tumors may also respond well to TKIs; however, the distinctions between these RCCs may warrant independent clinical trials to validate this as a therapeutic option. Clinical trials with FH-deficient tumors in HLRCC (AVATAR trial, NCT01130519) have shown promising results inhibiting angiogenesis by targeting the VEGF and EGFR pathways, which led to NCCN recommendation of this regimen in the treatment of HLRCC patients.35,36 One smaller study compared erlotinib/bevacizumab combinations to immunotherapy/ TKI combinations in FH-deficient RCC, reporting more favorable clinical outcomes in the latter. 37 Further molecular comparisons between responders and nonresponders may help to identify biomarkers that influence response to this treatment.
The most common co-mutation between FH-mut and WT tumors was in the NF2 gene (Figure 1), which prevents tumor growth through inhibition of different pathways including the RAS, PI3K/AKT, and HIPPO pathways.38,39 Concurrent mutations between FH and NF2 have also been previously reported in RCC.12,37,40 Previous studies have demonstrated that targeting YAP1 resulted in reduced tumor growth in NF2-mutated RCCs, suggesting that a significant subset of the FH-mut population may also benefit from targeting the Hippo pathway.41-43 Concurrent mutations were also observed between FH and DNA damage repair gene, BRCA1 (Figure 1). BRCA1 mutations are an indication for the use of Poly (ADP-ribose) polymerase (PARP) inhibitors in some tumor types. PARP inhibitors alone or in combination with other agents are also undergoing evaluation in VHLdeficient RCCs based on the DNA repair detect status and/or potential sensitivity of this genomic status to immunotherapies. 44-46 However, one phase II study determined the combination of PARP inhibitors and PD-L1 inhibitors to be ineffective in VHL-mutated RCC. 47 Accumulation of fumarate has also been reported to suppress the homologous recombination (HR) repair pathway, 48 which would not be detected by DNA sequencing, but suggests the sensitivity of FHmut tumors to DNA damaging agents. Co-mutations between FH and AKT1 were higher specifically in ccRCC. AKT signaling and the mTOR signaling pathway have also been linked to DNA damage response,49 suggesting that targeting these pathways may be useful for a subset of the population. Combinations of mTOR inhibitors and angiogenesis inhibitors have demonstrated robust activity in RCC patients.50 Other studies have used gene expression signatures to reveal prognostic value of angiogenesis signatures and T-cell-inflamed GEP signatures for use of VEGF and mTOR inhibitors in RCCs.51
RCCs have often been described as highly immunogenic, and numerous studies have demonstrated the efficacy of immunotherapies. In our study the analysis of immune checkpoint gene expression did not reveal significant differences between FH-mut and WT tumors. LAG3 exhibited the most significant difference in immune gene expression, which was observed specifically in non-ccRCC (Figure 3). Recent studies suggested that targeting LAG-3 might provide a promising partner in combinatorial immunotherapies in RCC,52 although our data may suggest differences between histology subtypes in this context. The analysis of immune cell infiltration also did not reveal significant changes between FHmut and WT tumors for most immune cells, with the exception of lower infiltration of NK cells and M2 macrophages in the non-RCC population (Figure 3). The IFN-γ response-related signature (IFN score) has been developed as a predictive biomarker for immunotherapy.53,54 When a similar IFN-γ response-related signature was described as a prognostic indicator in ccRCCs, a high-risk group exhibited low sensitivity to several drugs, not including immunotherapies.55 In our study, the IFN-γ responserelated signature was significantly reduced in FH-mut tumors in the non-ccRCC group, whereas the IFN score was higher in ccRCC (Figure 3). Although the development of biomarkers for stratification of patients in immunotherapy selection is ongoing in RCC, these results suggest that variability in histological subtypes might be considered in future studies. In our study, there were no significant differences observed between FH-mut and WT tumors for prevalence of TMB and MSI-H status (Figure 2). A previous study also reported low TMB and stable MSI status in both somatic and germline FH-mut RCCs.12 However, RCCs have been reported to have positive responses to immunotherapies and combination therapies with TKIs. A study of 336 ccRCC patients reported that higher TMB was correlated with lower immune cell infiltration and poor survival outcomes.56 Thus, contrary to what has been observed in some other cancer types, findings from numerous studies suggest that higher mutation rates in RCC are associated with immunologically cold tumor microenvironments. In contrast to TMB and MSI biomarkers, measurement of PD-L1 expression by IHC revealed higher expression of PD-L1 in FH-mut tumors, which was more pronounced in non-ccRCC (Figure 2). Several studies have described PD-L1 as a prognostic indicator of poor survival rates in immunotherapy-naive RCCs.57-59 Other studies have demonstrated the efficacy of ICI therapy for RCC, and the CheckMate 214 trial led to FDA approval of ICI combination therapy in 2018.60,61 However, most of these studies do not describe the genomic context to identify biomarkers that could differentiate responders from non-responders. In other tumor types, biomarkers developed to stratify patients for immunotherapy selection have included PD-L1 expression, TMB, MSI, and TME. However, the characteristics of RCCs have thus far precluded them from effective use of predictive ICI biomarkers. Follow-up studies from the CheckMate 214 trial reported that biomarkers previously associated with ICI benefit were not predictive in RCC.62 A few studies with small cohorts have analyzed drug response from FH-deficient RCCs with varying results. One study with 18 patients reported that patients with FHdeficient RCC who received immunotherapy had better clinical outcomes than patients who received antiangiogenic therapy alone.40 Conversely, another study with 24 patients reported that FHdeficient RCCs responded better to various antiangiogenics compared to immunotherapies or mTOR inhibitors.63 Therefore, further studies are needed to investigate the benefits of immunotherapies in FH-deficient renal cancers with the use of other potentially predictive biomarkers. Although a significant number of RCC patients respond to immunotherapy, a significant portion of this population also exhibits resistance, or develops resistance within several months. There is increasing interest in utilizing TKIs in combination with ICIs as a first- or second-line treatment, as well as understanding mechanisms of resistance.37,64,65 In addition to promoting angiogenesis and tumor migration, activation of HIF and increased VEGF also influence changes in the tumor microenvironment, including the release of immunosuppressive factors such as PD-L1.34 Alternately, reports have described that antiangiogenic molecules can overcome immunosuppressive networks and thereby influence sensitivity of tumors to immunotherapies.66 Analyzing the molecular features of FH-deficient RCCs in the context of drug response will be beneficial for better understanding the clinical and therapeutic implications of such a dynamic system.
Study Limitations
The primary limitation of this study is the small cohort size. Although it is the largest cohort of FH-deficient RCC tumors that has been described by comprehensive molecular profiling, a total sample size of 40 FH-mut samples limits the comparative analyses that could be performed. Although there is considerable heterogeneity among RCCs, small cohort size also limits conclusions about prevalence in histological subtypes, molecular subtypes, or comparisons between primary and metastatic tumors. Furthermore, this study does not include other variables which may impact the molecular landscape of the sequenced samples, including demographic information, or clinical information about staging or treatment regimens. The lack of clinical outcomes data associated with the cohort also limits the clinical implications that can be drawn from the observed molecular characteristics. RNA-Seq data from FFPE samples also has inherent limitations which should be considered with immune profiling methods used in this study. Despite the rare incidence of FH-mut RCCs, the accurate diagnosis of FH status is important due to their aggressive nature. The accumulation of succinate dehydrogenase in FHdeficient tumors provides a highly immunoreactive target in recently improved IHC-based screening methods that could provide clinical utility for many renal cancers.67,68 In conclusion, comprehensive genomic profiling of FH-deficient RCCs yielded results in this study that were generally consistent with prior reports of genetic this difference wacharacteristics of FH-deficient RCCs, while analysis of a large sample size enabled observation of some interesting trends. FHdeficient RCCs exhibit molecular characteristics that may distinguish them from WT-RCCs. Common RCC driver mutations of renal cell carcinoma (RCC) are reduced in FH-mutated RCC, while some other co-mutations are increased that may point to new therapeutic options. Overall, these findings point to several possible trends in FH-deficient RCC that could be validated in a larger clinical study, while comprehensive molecular analysis may provide the potential for identifying novel therapeutic approaches in FH-deficient RCC.
SUPPLEMENTAL INFORMATION
Access supplemental information online. https://kidney-cancerjournal. com/KCJ21n4-or1.php
AUTHORS DISCLOSURES:
SW, AH, AG, AH, and CN are employees of Caris Life Sciences. BN reports a consulting or advisory role for Exelixis. PCB reports a consulting or advisory role for Bayer, BMS, Caris Life Sciences, Clovis Oncology, Dendreon, Eisai, EMD Serono, and Pfizer; PCB reports research funding from Blue Earth Diagnostics. CJR reports a consulting or advisory role for Advanced Accelerator Applications, Bayer, Clovis Oncology, Dendreon, Myovant Sciences, and Roivant; CJR reports receiving honoraria from Bayer and Janssen Oncology; CJR reports research funding from Clovis Oncology and Genzyme. RRM reports a consulting or advisory role for Astellas Medivation, AstraZeneca, Bayer, Bristol-Myers Squibb, Calithera Biosciences, Caris Life Sciences, Dendreon, Exelixis, Janssen, Merck, Myovant Sciences, Novartis, Pfizer, Sanofi, Sorrento Therapeutics, Tempus, and Vividion Therapeutics; RRM reports research funding from Bayer, Pfizer, and Tempus. EIH reports a consulting or advisory role for Agensys, AstraZeneca, Bayer, Dendreon, and Sanofi; EIH reports receiving honoraria from AstraZeneca, Bayer, Dendreon, Sanofi, and Seattle Genetics. EIH reports funding for travel and/or the speaker’s bureau from Sanofi, Agensys, and Bayer; EIH reports research funding from Agensys, AIQ Solutions, Astellas Pharma, AstraZeneca, AstraZeneca, Bayer, Boehringer Ingelheim, Bristol-Myers Squibb, Calibr, Caris Life Sciences, Celgene, Celldex, Champions Oncology, Corcept Therapeutics, Curemeta, Daiichi Sankyo Inc., Dendreon, eFFECTOR Therapeutics, Eisai, Esanik, Five Prime Therapeutics, Fortis, Genentech/Roche, GlaxoSmithKline, Ignyta, Infinity Pharmaceuticals, Inovio Pharmaceuticals, Janssen Research & Development, Medivation, Merck, Merck Sharp & Dohme, Millennium, Mirati Therapeutics, Modra Pharmaceuticals, Novartis, Oncolys BioPharma, Pellficure, Peloton Therapeutics, Pharmacyclics, Plexxikon, Seattle Genetics, Synta, Tokai Pharmaceuticals, Zenith Epigenetics, and Zenith Epigenetics. All other authors have declared no conflicts of interest.


REFERENCE
1. Tomlinson IP, Alam NA, Rowan AJ,
et al. Germline mutations in FH predispose
to dominantly inherited uterine fibroids,
skin leiomyomata and papillary renal cell
cancer. Nat Genet 2002;30:406-10.
2. Wei MH, Toure O, Glenn GM, et
al. Novel mutations in FH and expansion
of the spectrum of phenotypes expressed in
families with hereditary leiomyomatosis and
renal cell cancer. J Med Genet 2006;43:18-
27.
3. Isaacs JS, Jung YJ, Mole DR, et al.
HIF overexpression correlates with biallelic
loss of fumarate hydratase in renal cancer:
novel role of fumarate in regulation of HIF
stability. Cancer Cell 2005;8:143-53.
4. Pollard PJ, Brière JJ, Alam
NA, et al. Accumulation of Krebs cycle
intermediates and over-expression of
HIF1alpha in tumours which result from
germline FH and SDH mutations. Hum Mol
Genet 2005;14:2231-9.
5. Xiao M, Yang H, Xu W, et al.
Inhibition of α-KG-dependent histone
and DNA demethylases by fumarate and
succinate that are accumulated in mutations
of FH and SDH tumor suppressors. Genes
Dev 2012;26:1326-38.
6. Grubb RL, 3rd, Franks ME, Toro J,
et al. Hereditary leiomyomatosis and renal
cell cancer: a syndrome associated with an
aggressive form of inherited renal cancer. J
Urol 2007;177:2074-9; discussion 9-80.
7. Launonen V, Vierimaa O, Kiuru
M, et al. Inherited susceptibility to uterine
leiomyomas and renal cell cancer. Proc Natl
Acad Sci U S A 2001;98:3387-92.
8. Toro JR, Nickerson ML, Wei MH, et
al. Mutations in the fumarate hydratase gene
cause hereditary leiomyomatosis and renal
cell cancer in families in North America. Am
J Hum Genet 2003;73:95-106.
9. Forde C, Lim DHK, Alwan Y, et al.
Hereditary Leiomyomatosis and Renal Cell
Cancer: Clinical, Molecular, and Screening
Features in a Cohort of 185 Affected
Individuals. Eur Urol Oncol 2020;3:764-72.
10. Chayed Z, Kristensen LK, Ousager
LB, Rønlund K, Bygum A. Hereditary
leiomyomatosis and renal cell carcinoma: a
case series and literature review. Orphanet J
Rare Dis 2021;16:34.
11. Menko FH, Maher ER, Schmidt
LS, et al. Hereditary leiomyomatosis and
renal cell cancer (HLRCC): renal cancer risk,
surveillance and treatment. Fam Cancer
2014;13:637-44.
12. Gleeson JP, Nikolovski I, Dinatale
R, et al. Comprehensive Molecular
Characterization and Response to Therapy
in Fumarate Hydratase-Deficient Renal Cell
Carcinoma. Clin Cancer Res 2021;27:2910-
9.
13. Kiuru M, Lehtonen R, Arola J, et al.
Few FH mutations in sporadic counterparts
of tumor types observed in hereditary
leiomyomatosis and renal cell cancer
families. Cancer Res 2002;62:4554-7.
14. Thouvenin J, Masson C, Boudier P,
et al. Complete Response in Metastatic Clear
Cell Renal Cell Carcinoma Patients Treated
with Immune-Checkpoint Inhibitors:
Remission or Healing? How to Improve
Patients' Outcomes? Cancers (Basel)
2023;15.
15. Marabelle A, Fakih M, Lopez J,
et al. Association of tumour mutational
burden with outcomes in patients with
advanced solid tumours treated with
pembrolizumab: prospective biomarker
analysis of the multicohort, open-label,
phase 2 KEYNOTE-158 study. Lancet Oncol
2020;21:1353-65.
16. Merino DM, McShane LM,
Fabrizio D, et al. Establishing guidelines to
harmonize tumor mutational burden (TMB):
in silico assessment of variation in TMB
quantification across diagnostic platforms:
phase I of the Friends of Cancer Research
TMB Harmonization Project. J Immunother
Cancer 2020;8.
17. Finotello F, Mayer C, Plattner
C, et al. Molecular and pharmacological
modulators of the tumor immune contexture
revealed by deconvolution of RNA-seq data.
Genome Med 2019;11:34.
18. Cristescu R, Mogg R, Ayers M, et
al. Pan-tumor genomic biomarkers for PD-1
checkpoint blockade-based immunotherapy.
Science 2018;362.
19. Dall'Oglio MF, Antunes AA,
Pompeo AC, Mosconi A, Leite KR, Srougi
M. Prognostic relevance of the histological
subtype of renal cell carcinoma. Int Braz J
Urol 2008;34:3-8.
20. Keegan KA, Schupp CW, Chamie
K, Hellenthal NJ, Evans CP, Koppie TM.
Histopathology of surgically treated renal
cell carcinoma: survival differences by
subtype and stage. J Urol 2012;188:391-7.
21. Leibovich BC, Lohse CM,
Crispen PL, et al. Histological subtype is
an independent predictor of outcome for
patients with renal cell carcinoma. J Urol
2010;183:1309-15.
22. Muglia VF, Prando A. Renal cell
carcinoma: histological classification and
correlation with imaging findings. Radiol
Bras 2015;48:166-74.
23. Miao D, Margolis CA, Gao W, et al.
Genomic correlates of response to immune
checkpoint therapies in clear cell renal cell
carcinoma. Science 2018;359:801-6.
24. Motzer RJ, Banchereau R, Hamidi
H, et al. Molecular Subsets in Renal
Cancer Determine Outcome to Checkpoint
and Angiogenesis Blockade. Cancer Cell
2020;38:803-17.e4.
25. Jee B, Seo E, Park K, et al.
124 Kidney Cancer Journal | 21 (4) DEC 2023 Kidney-Cancer-Journal.com
Molecular Subtypes Based on Genomic and
Transcriptomic Features Correlate with the
Responsiveness to Immune Checkpoint
Inhibitors in Metastatic Clear Cell Renal Cell
Carcinoma. Cancers (Basel) 2022;14.
26. Gnarra JR, Tory K, Weng Y, et al.
Mutations of the VHL tumour suppressor
gene in renal carcinoma. Nat Genet
1994;7:85-90.
27. Sudarshan S, Shanmugasundaram
K, Naylor SL, et al. Reduced expression
of fumarate hydratase in clear cell renal
cancer mediates HIF-2α accumulation and
promotes migration and invasion. PLoS One
2011;6:e21037.
28. Chakraborty S, Balan M,
Sabarwal A, Choueiri TK, Pal S. Metabolic
reprogramming in renal cancer: Events of
a metabolic disease. Biochim Biophys Acta
Rev Cancer 2021;1876:188559.
29. Stransky LA, Vigeant SM, Huang
B, et al. Sensitivity of VHL mutant kidney
cancers to HIF2 inhibitors does not require
an intact p53 pathway. Proc Natl Acad Sci U
S A 2022;119:e2120403119.
30. Choueiri TK, Halabi S, Sanford
BL, et al. Cabozantinib Versus Sunitinib As
Initial Targeted Therapy for Patients With
Metastatic Renal Cell Carcinoma of Poor or
Intermediate Risk: The Alliance A031203
CABOSUN Trial. J Clin Oncol 2017;35:591-
7.
31. Motzer RJ, Hutson TE, Cella D, et
al. Pazopanib versus sunitinib in metastatic
renal-cell carcinoma. N Engl J Med
2013;369:722-31.
32. Motzer RJ, Hutson TE, Tomczak
P, et al. Overall survival and updated results
for sunitinib compared with interferon
alfa in patients with metastatic renal cell
carcinoma. J Clin Oncol 2009;27:3584-90.
33. Sternberg CN, Davis ID, Mardiak
J, et al. Pazopanib in locally advanced or
metastatic renal cell carcinoma: results of
a randomized phase III trial. J Clin Oncol
2010;28:1061-8.
34. Mennitto A, Huber V, Ratta R, et
al. Angiogenesis and Immunity in Renal
Carcinoma: Can We Turn an Unhappy
Relationship into a Happy Marriage? J Clin
Med 2020;9.
35. NCCN Guidelines-Kidney Cancer.
NCCN, 2022. (Accessed 2022/11/10,
at https://www.nccn.org/guidelines/
guidelines-detail?category=1&id=1440.)
36. Srinivasan R, Gurram S, Harthy
MA, et al. Results from a phase II study
of bevacizumab and erlotinib in subjects
with advanced hereditary leiomyomatosis
and renal cell cancer (HLRCC) or sporadic
papillary renal cell cancer. Journal of Clinical
Oncology 2020;38:5004-.
37. Xu Y, Kong W, Cao M, et al.
Genomic Profiling and Response to Immune
Checkpoint Inhibition plus Tyrosine Kinase
Inhibition in FH-Deficient Renal Cell
Carcinoma. Eur Urol 2022.
38. Dey A, Varelas X, Guan KL.
Targeting the Hippo pathway in cancer,
fibrosis, wound healing and regenerative
medicine. Nat Rev Drug Discov 2020;19:480-
94.
39. Petrilli AM, Fernández-Valle C.
Role of Merlin/NF2 inactivation in tumor
biology. Oncogene 2016;35:537-48.
40. Sun G, Zhang X, Liang J, et al.
Integrated Molecular Characterization of
Fumarate Hydratase-deficient Renal Cell
Carcinoma. Clin Cancer Res 2021;27:1734-
43.
41. Calses PC, Crawford JJ, Lill JR,
Dey A. Hippo Pathway in Cancer: Aberrant
Regulation and Therapeutic Opportunities.
Trends Cancer 2019;5:297-307.
42. Sourbier C, Liao PJ, Ricketts CJ,
et al. Targeting loss of the Hippo signaling
pathway in NF2-deficient papillary kidney
cancers. Oncotarget 2018;9:10723-33.
43. White SM, Avantaggiati ML,
Nemazanyy I, et al. YAP/TAZ Inhibition
Induces Metabolic and Signaling Rewiring
Resulting in Targetable Vulnerabilities
in NF2-Deficient Tumor Cells. Dev Cell
2019;49:425-43.e9.
44. Ged Y, Chaim JL, DiNatale RG, et
al. DNA damage repair pathway alterations
in metastatic clear cell renal cell carcinoma
and implications on systemic therapy. J
Immunother Cancer 2020;8.
45. Pletcher JP, Bhattacharjee S, Doan
JP, et al. The Emerging Role of Poly (ADPRibose)
Polymerase Inhibitors as Effective
Therapeutic Agents in Renal Cell Carcinoma.
Front Oncol 2021;11:681441.
46. Scanlon SE, Hegan DC, Sulkowski
PL, Glazer PM. Suppression of homologydependent
DNA double-strand break repair
induces PARP inhibitor sensitivity in VHLdeficient
human renal cell carcinoma.
Oncotarget 2018;9:4647-60.
47. Kotecha R, Lee C-H, McHugh DJ,
et al. A phase II study of talazoparib and
avelumab in VHL deficient clear cell renal
cell carcinoma. Journal of Clinical Oncology
2022;40:347-.
48. Sulkowski PL, Sundaram RK, Oeck
S, et al. Krebs-cycle-deficient hereditary
cancer syndromes are defined by defects in
homologous-recombination DNA repair.
Nat Genet 2018;50:1086-92.
49. Ma Y, Vassetzky Y, Dokudovskaya
S. mTORC1 pathway in DNA damage
response. Biochim Biophys Acta Mol Cell
Res 2018;1865:1293-311.
50. Feldman DR, Ged Y, Lee CH,
et al. Everolimus plus bevacizumab is an
effective first-line treatment for patients
with advanced papillary variant renal cell
carcinoma: Final results from a phase II
trial. Cancer 2020;126:5247-55.
51. Donskov F, Pinto CA, Predoiu
R, et al. Molecular analysis and favorable
clinical outcomes in real-world patients with
metastatic renal cell carcinoma. Acta Oncol
2022;61:1268-77.
52. Zelba H, Bedke J, Hennenlotter J,
et al. PD-1 and LAG-3 Dominate Checkpoint
Receptor-Mediated T-cell Inhibition in
Renal Cell Carcinoma. Cancer Immunol Res
2019;7:1891-9.
53. Ayers M, Lunceford J, Nebozhyn
M, et al. IFN-γ-related mRNA profile
predicts clinical response to PD-1 blockade.
J Clin Invest 2017;127:2930-40.
54. Cui C, Xu C, Yang W, et al.
Ratio of the interferon-γ signature to the
immunosuppression signature predicts anti-
PD-1 therapy response in melanoma. NPJ
Genom Med 2021;6:7.
55. Liu L, Du X, Fang J, et al.
Development of an Interferon Gamma
Response-Related Signature for Prediction of
Survival in Clear Cell Renal Cell Carcinoma.
J Inflamm Res 2021;14:4969-85.
56. Zhang C, Li Z, Qi F, Hu X, Luo J.
Exploration of the relationships between
tumor mutation burden with immune
infiltrates in clear cell renal cell carcinoma.
Ann Transl Med 2019;7:648.
57. Lu Y, Song Y, Xu Y, et al.
The prevalence and prognostic and
clinicopathological value of PD-L1 and
PD-L2 in renal cell carcinoma patients:
a systematic review and meta-analysis
involving 3,389 patients. Transl Androl Urol
2020;9:367-81.
58. Möller K, Fraune C, Blessin NC,
et al. Tumor cell PD-L1 expression is a
strong predictor of unfavorable prognosis
in immune checkpoint therapy-naive clear
cell renal cell cancer. Int Urol Nephrol
2021;53:2493-503.
59. Ueda K, Suekane S, Kurose H,
et al. Prognostic value of PD-1 and PDL1
expression in patients with metastatic
clear cell renal cell carcinoma. Urol Oncol
2018;36:499.e9-.e16.
60. Motzer RJ, Escudier B, McDermott
DF, et al. Nivolumab versus Everolimus in
Advanced Renal-Cell Carcinoma. N Engl J
Med 2015;373:1803-13.
61. Motzer RJ, Tannir NM, McDermott
DF, et al. Nivolumab plus Ipilimumab
versus Sunitinib in Advanced Renal-Cell
Carcinoma. N Engl J Med 2018;378:1277-
90.
62. Motzer RJ, Choueiri TK,
McDermott DF, et al. Biomarker analysis
from CheckMate 214: nivolumab plus
ipilimumab versus sunitinib in renal cell
carcinoma. J Immunother Cancer 2022;10.
63. Carril-Ajuria L, Colomba E, Cerbone
L, et al. Response to systemic therapy in
fumarate hydratase-deficient renal cell
carcinoma. Eur J Cancer 2021;151:106-14.
64. Sharma R, Kadife E, Myers M,
Kannourakis G, Prithviraj P, Ahmed N.
Determinants of resistance to VEGF-TKI and
immune checkpoint inhibitors in metastatic
renal cell carcinoma. J Exp Clin Cancer Res
2021;40:186.
65. Vano YA, Ladoire S, Elaidi R, et
al. First-Line Treatment of Metastatic Clear
Cell Renal Cell Carcinoma: What Are the
Most Appropriate Combination Therapies?
Cancers (Basel) 2021;13.
66. Kwilas AR, Donahue RN, Tsang
KY, Hodge JW. Immune consequences of
tyrosine kinase inhibitors that synergize
with cancer immunotherapy. Cancer Cell
Microenviron 2015;2.
67. Trpkov K, Siadat F.
Immunohistochemical screening for the
diagnosis of succinate dehydrogenase deficient
renal cell carcinoma and fumarate
hydratase-deficient renal cell carcinoma.
Ann Transl Med 2019;7:S324.
68. Gupta S, Swanson AA, Chen YB,
et al. Incidence of succinate dehydrogenase
and fumarate hydratase-deficient renal cell
carcinoma based on immunohistochemical
screening with SDHA/SDHB and FH/2SC.
Hum Pathol 2019;91:114-22