


Renal Cell Carcinoma Associated with Germline Mutations in the Krebs Cycle
Eric Lu 1, Brian Shuch,2, Alexandra Drakaki1,2 1 Division of Hematology/Oncology, University of California, Los Angeles, Los Angeles, CA, USA; 2 Institute of Urologic Oncology, University of California Los Angeles, Los Angeles, CA, USA;ABSTRACT
Germline mutations in the fumarate hydratase (FH) and succinate dehydrogenase (SDH) genes lead to hereditary leiomyomatosis and RCC (HLRCC) and hereditary paraganglioma and pheochromocytoma, respectively. The renal cell carcinomas that arise in these conditions are characterized by dysregulated Krebs cycles, accumulation of oncometabolites, downstream changes in gene expression, and epigenetic modifications that carry unique therapeutic implications. In this review, we evaluate the current literature on these tumors, including the epidemiology, clinical course, screening guidelines, and management of localized and metastatic disease.
INTRODUCTION
Renal cell carcinoma (RCC) is one of the most common cancers in the United States, with approximately 72,000 new cases and 13,000 deaths projected for 20211. While most RCC occurs sporadically, there is an increasing recognition of the hereditary component for a subset of patients. It is believed that approximately 5-8% of RCC has a strong hereditary component; however, heritability is complex, and kidney cancer genetic predisposition may extend far beyond monogenetic diseases2,3. Current estimates, while likely biased, do reinforce that these estimates are not too far off; for instance, large studies such as The Cancer Genome Atlas Program (TCGA) have reported that clear cell, papillary, and chromophobe RCC are associated with germline pathogenic mutations in 6%, 9%, and 6% of individuals respectively. 4 Single-institution series have reported higher numbers of pathogenic germline mutations (up to 16%) with further enrichment in non-clear cell variants. However, over two-thirds of these mutations did not occur in classic RCC genes, suggesting that they are potentially unrelated or, alternatively, that we have yet to characterize the full spectrum of kidney cancer predisposition5.
Several autosomal dominant RCC syndromes and associated germline mutations have been well-described, including von Hippel-Lindau (VHL gene), hereditary papillary renal cell carcinoma (MET gene), Birt-Hogg- Dube´ (FLCN gene), hereditary leiomyomatosis and RCC (FH gene), succinate dehydrogenase (SDH) deficient kidney cancer (SDHA, SDHB, SDHC, SDHD genes), tuberous sclerosis complex (TSC1 and TSC2 genes), Cowden syndrome (PTEN gene), and microphthalmia- associated transcription factor kidney cancer (MITF gene)6.
Herein, we will focus on hereditary leiomyomatosis and RCC (HLRCC) and SDH-deficient kidney cancer (also known as hereditary paraganglioma and pheochromocytoma), as these two conditions are characterized by Krebs cycle dysfunction, accumulation of oncometabolites, and subsequent predisposition of affected individuals to various malignancies6. This link between mitochondrial physiology and tumorigenesis was first observed in the 1920s by Otto Warburg7 Accordingly, the genes encoding the Krebs cycle enzymes fumarate hydratase (FH) and the succinate dehydrogenase complex (SDHA, SDHB, SDHC, SDHD as well as SDHAF2 which encodes a protein essential for complex assembly) act as tumor suppressors, with germline mutations in these genes leading to accumulation of fumarate and succinate, respectively8 These deficits in mitochondrial respiration have been linked to tumorigenesis via several proposed mechanisms. Accumulation of fumarate and succinate as oncometabolites has been shown to inhibit the degradation of HIF1, leading to a state of pseudohypoxia and subsequent downstream gene expression leading to tumorigenesis. Concurrently, excessive fumarate and succinate accumulation leads to DNA and histone methylation, resulting in altered gene expression via epigenetic silencing9-11. More recent work has demonstrated that oncometabolite- induced disruption of chromatin signaling leads to an intrinsic homologous recombination DNA repair defect and perhaps further mutational burden. Such findings suggest that FHand SDH-deficient RCC may represent unique forms of kidney cancer that require different therapeutic approaches.
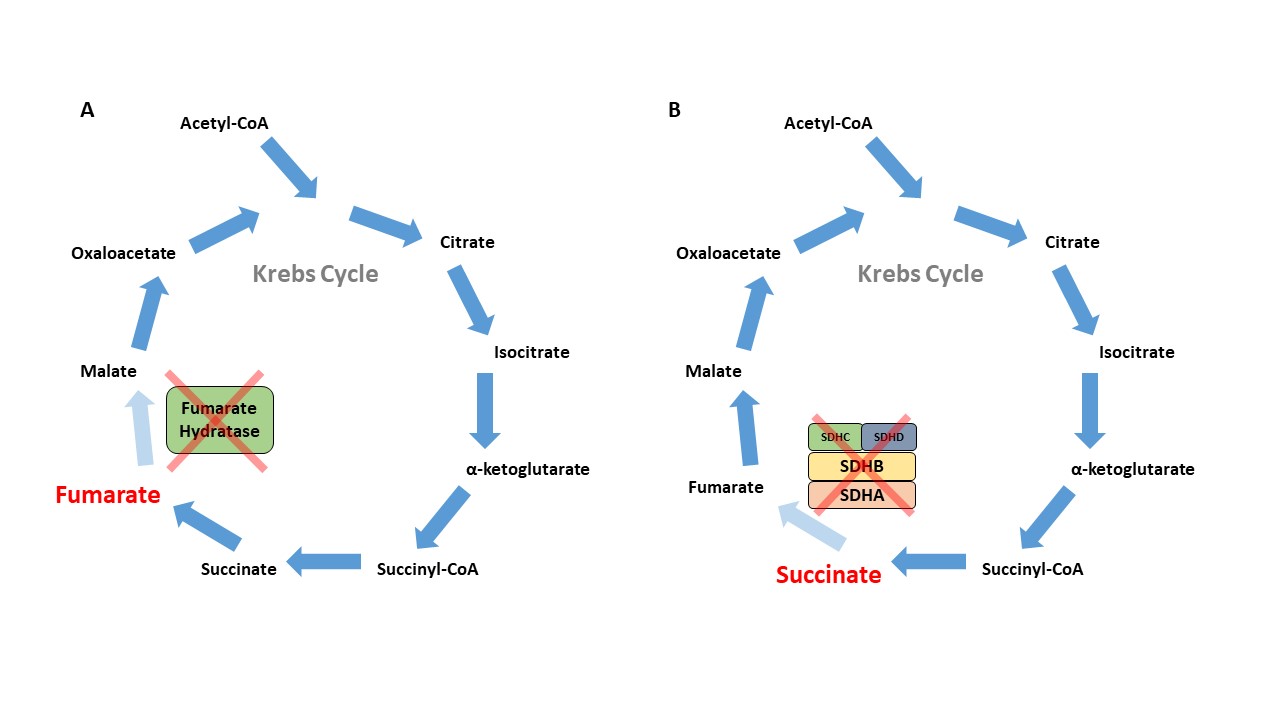
HLRCC
The FH gene encodes for the Krebs cycle enzyme fumarate hydratase, which catalyzes the conversion of fumarate to malate (Figure 1A). The loss of function in the FH gene leads to the autosomal dominant cancer syndrome known as HLRCC, which is characterized by cutaneous leiomyomas, early-onset and highly symptomatic uterine leiomyomas, adrenal macronodular hyperplasia, and a very aggressive form of kidney cancer now recognized as its own subtype – FH-deficient kidney cancer. This subtype can resemble papillary type 2, collecting duct, and tubulocystic RCC (Figure 2)13,14. The incidence and prevalence of HLRCC are currently unknown, but now several hundred families have been described in the literature. 15. The prevailing consensus was that HLRCC is quite rare, and among those with pathogenic FH mutations, the lifetime cumulative risk of RCC was 15-30%16,17. However with the widespread availability of panel testing that included the FH gene, many more patients are now being identified, leading to the belief that this condition is under- recognized. With large exome databases available, it is now evident that FH alterations are very common with carrier estimates between 1/1000 and 1/2500 individuals, suggesting a much lower RCC penetrance closer to 2-6%18.
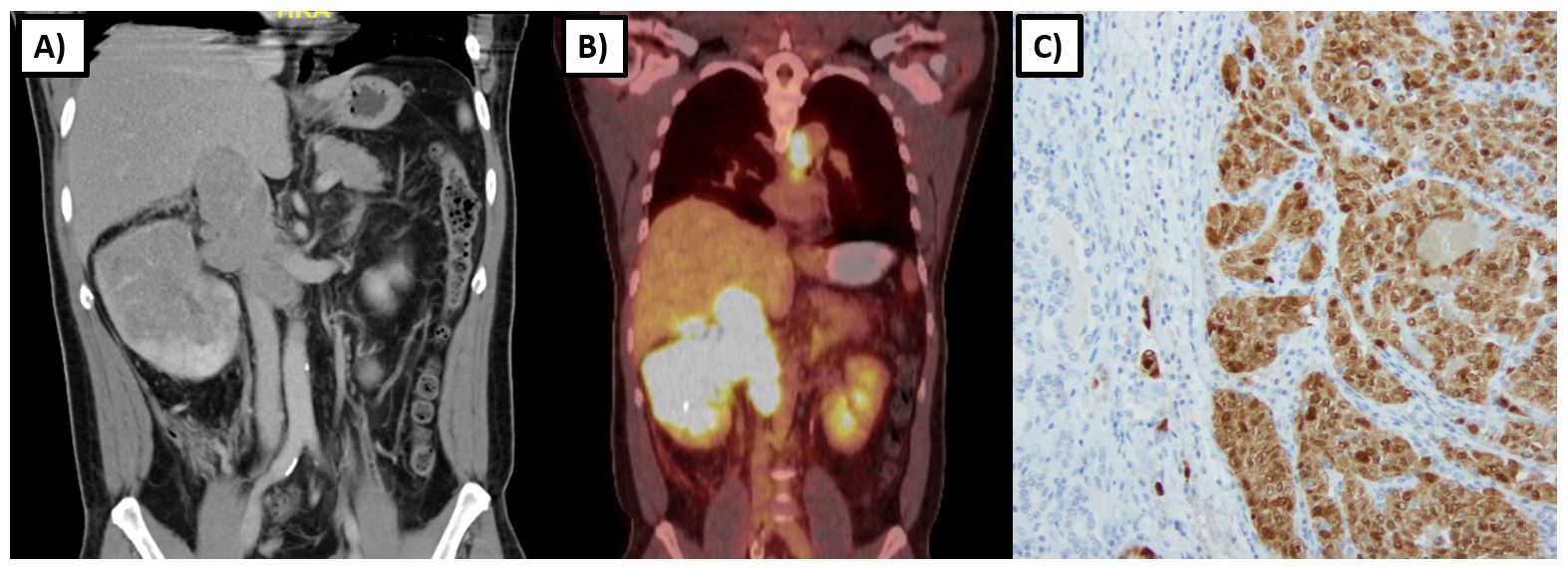
Renal tumors in HLRCC tend to develop at an earlier age, with one series reporting a median age of onset of 37 years with a range of 10-773. Tumors tend to be unilateral and solitary with a particularly aggressive biological behavior compared to other types of hereditary kidney cancer. Imaging characteristics frequently demonstrate an infiltrative nature (>85%) with the invasion of the renal sinus fat (>80%) (Figure 2)19. Even smaller tumors have a propensity for early and rapid nodal and distant metastasis, as evidenced by one series in which four of seven patients with 2.0-6.7 cm T1 tumors had spread to regional lymph nodes or had distant metastases at the time of nephrectomy. 20 Another study found that among HLRCC patients with RCC, 47% (16/34) were metastatic at diagnosis, and another 35% (12/34) became metastatic within 3 years of diagnosis21.
Due to this accelerated growth rate and potential for metastatic spread even with small primary tumors, annual abdominal imaging (MRI favored over CT) is recommended for surveillance starting at age 8-10 for those at risk16,22. For localized tumors, surveillance is not recommended, even for smaller tumors less than 1 cm. Rather, partial nephrectomy with wide surgical margins with consideration of retroperitoneal lymphadenectomy is recommended. Radical nephrectomy could also be considered if partial nephrectomy is not felt to be able to achieve a wide margin.16 Although enucleation for small RCC tumors has increased in popularity over recent years, for FH-deficient RCC, the surgeon must keep a safe distance away from the tumor as local recurrences are common.
For metastatic HLRCC patients, therapeutic options are unfortunately limited with no accepted standard. Given that oncometabolite (fumarate) accumulation leads to a disruption of the Krebs cycle and a shift towards dependence on aerobic glycolysis for energy needs, these tumors may become uniquely dependent on aerobic glycolysis, which is sustained by high glucose influx. Furthermore, tumorigenesis is driven by the pseudohypoxia pathway, rendering these tumors particularly sensitive to drugs directed against molecular targets downstream of HIF1-alpha. Consequentially, vascular endothelial growth factor receptor (VEGF) pathway inhibitors represent the most rational therapeutic choice over immunotherapy.
Several ongoing clinical trials are testing rational drug targets in patients with HLRCC-associated renal tumors. The combination of bevacizumab plus erlotinib has shown promising activity in an ongoing phase II clinical trial of patients with papillary RCC, both HLRCC and sporadic (NCT01130519). Results from an abstract published in May of 2020 detail that for the 83 treated patients, the objective response rate was 64% (27/42) in the HLRCC cohort and 37% (15/41) in a sporadic papillary RCC cohort. Median PFS was 21.1 months in the HLRCC cohort and 8.7 months in the sporadic cohort23. Most adverse events were grade 1-2, and the most notable grade ≥3 adverse events were hypertension (34%) and proteinuria (13%), as expected from bevacizumab. In addition, one patient died from gastrointestinal hemorrhage possibly attributable to bevacizumab therapy. The investigators postulated that the increased activity among HLRCC patients may be related to FH inactivation resulting in upregulation of HIF, with the resulting metabolic alterations leading the tumors to be uniquely susceptible to this combination.
The responsiveness to this regimen was also observed in a small retrospective series from South Korea where the objective response rate was 50% (5/10)24. A more recent randomized phase 2 trial, SWOG 1500, established cabozantinib (a dual MET and VEGF inhibitor) as a promising option for patients with papillary RCC. However, given that HLRCC-associated renal tumors (previously classified as papillary type 2) represent a distinct morphologic and molecular subset that is metabolically deficient, rather than MET-driven as with papillary tumors, it remains unclear whether the results from this trial are generalizable to HLRCC patients. Nonetheless, for the small subset of patients in this trial with HLRCC, it will be important to follow the long-term outcomes on cabozantinib14,25.
Other strategies involving immunotherapy have been described in case reports. In one instance, the use of axitinib plus the PD-1 blocker sintilimab in a patient with metastatic HLRCC-associated RCC resulted in disease stabilization and improvement of symptoms26. In another case, combination immunotherapy with nivolumab plus ipilimumab for metastatic disease led to a complete response with an ongoing durable remission at 68 weeks27. However, given that larger studies are lacking, it remains unclear at this time what role immunotherapy plays in the treatment of this disease22.
SDH-DEFICIENT RCC
SDH is an enzyme complex that is composed of four subunits (SDHA, SDHB, SDHC, SDHD) and assembled with SDHAF1 and SDHAF2 mitochondrial proteins. This enzyme plays a critical role in mitochondrial respiration; specifically, it catalyzes the oxidation of succinate to fumarate within the Krebs cycle and is also critical to complex 2 of the electron transport chain (Figure 1B). Those with germline loss of function mutations in one of these SDH genes are at risk for paragangliomas, pheochromocytomas, gastrointestinal stromal tumors (GIST), and RCC.28 RCC has been reported in individuals with SDHB, SDHC, and SDHD mutations, and more recently, in patients with SDHA mutations29. Histology can be variable and may depend on the subunit affected. SDHB-deficient tumors are characterized by a unique oncocytic and vacuolated appearance. By contrast, SDHC-deficient tumors tend to present with clear cell histology28,30. Various other histologies, including papillary, sarcomatoid, and unclassified RCCs have been reported in patients with germline mutations of SDH subunit genes (Figure 3).
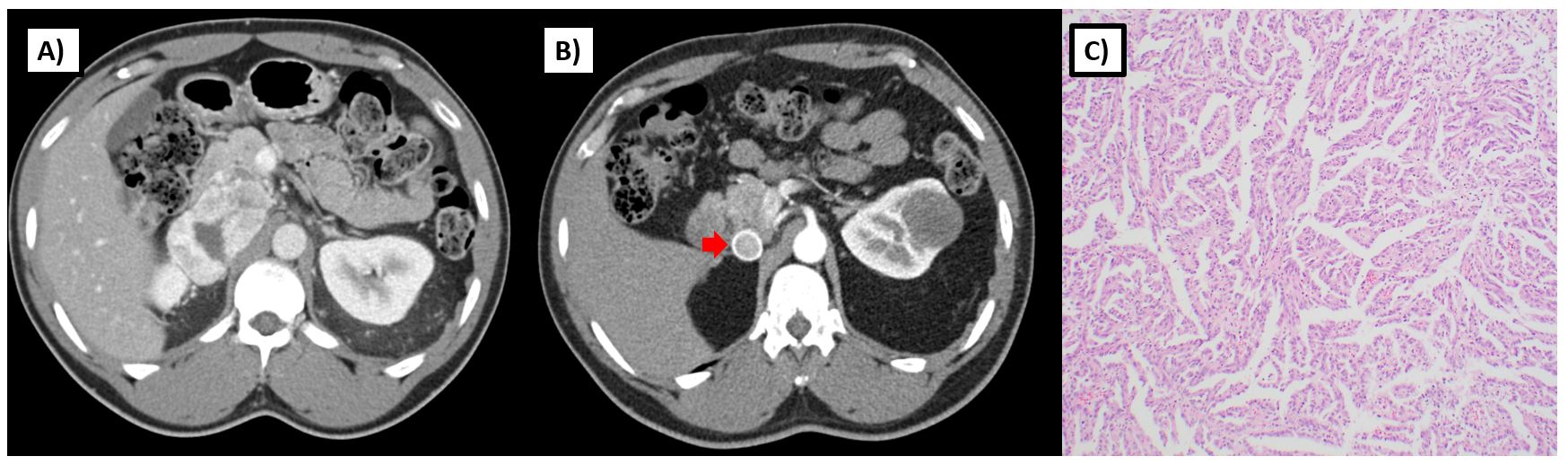
As opposed to sporadic RCC, SDH-deficient RCC tends to occur at an earlier age with a reported median age of onset 30 years and range from 15 to 61 in one series3. Given that these tumors share common underlying metabolic features with FH-deficient RCC, some may behave in a similarly aggressive manner and present with metastatic disease; however, others may have a more low-grade appearance and present with non-invasive tumors (Figure 3). Although penetrance for paragangliomas and pheochromocytomas are much higher in those with a germline SDH mutation (18-95% by age 60), RCC can also present as the sole finding in such individuals28,31. The true penetrance for RCC in SDH-deficiency is unknown and may vary by the affected subunit, but some reports estimate that risk could be up to 14% by age 7032. Given the variable histologies and uncertain penetrance of RCC in SDH-deficiency, screening recommendations vary. The National Comprehensive Cancer Network recommends abdominal imaging (MRI or CT) with and without contrast every 4-6 years starting at age 1222. However this approach should be individualized based on family risk.
SDH-deficient RCC tends to have a lower risk for metastatic disease but a high incidence of bilateral tumors (with 26% bilateral tumors in one series of 27 patients with prolonged up). Nonetheless, surveillance, even for smaller tumors, is not recommended32. For individuals with non-invasive tumors, nephron-sparing surgery can be considered. However, for individuals with higher risk tumors (large, invasive, high-grade, infiltrative), the risk for development of metastatic disease is high, and radical nephrectomy with lymph node dissection should be considered33. Patients will require longterm follow-up due to potential for late recurrences, metachronous disease, and other syndromic manifestations of germline SDH deficiency (ie, paraganglioma, pheochromocytomas, GIST).
For metastatic SDH-deficient RCC, there is no widely accepted frontline therapy, as evidence is limited to single case series. Much like HLRCC, given the unique metabolic disturbance caused by oncometabolite (succinate) accumulation, VEGF inhibitors are commonly used. One case of widely metastatic RCC of unclassifiable histology with high-grade features was initially treated with sunitinib with a 15-month duration of response, followed by temsirolimus with only a 2-month duration of response, before ultimately being found to be positive for a SDHB mutation. The patient was subsequently treated with pazopanib (a multi-kinase angiogenesis inhibitor) with symptomatic response and stabilization of metastatic disease. However, the patient experienced progression of his cancer 6 months later and ultimately succumbed to the disease. 34 In another report, an individual with SDHC-deficient clear cell RCC who eventually developed widespread metastases not amenable to locally directed therapies was treated with sunitinib and remained on therapy for 34 months with a near-complete response35. In a third case, an individual with a SDHA germline mutation developed a highgrade papillary type 2 RCC with sarcomatoid dedifferentiation that was initially refractory to anti-PD-1 treatment, but later experienced disease stabilization while on a series of VEGF tyrosine kinase inhibitors (sunitinib, pazopanib, and sorafenib)36.
CONCLUSIONS
Although accounting for a minority of RCC cases, Krebs cycle deficient RCC represents a molecularly distinct subset that is often extremely clinically aggressive. Given the complexities of diagnosis and management of such diseases, a multi-disciplinary approach is critical. For now, early surgical excision is the standard for localized disease, and VEGF-based therapy remains the mainstay for metastatic disease. However, moving forward, the unique metabolic features of Krebs cycle deficient tumors may be amenable to more refined therapeutic targeting, and many such efforts are already underway to improve clinical outcomes.
REFERENCES
KEYWORDS: Krebs Cycle • Germline Mutations • Oncometabolites • Renal Cell Carcinoma • HLRCC • Papillary RCC
Correspondence:Alexandra Drakaki, MD, PhD. Division of Hematology/Oncology and Institute of Urologic Oncology UCLA Health. adrakaki@mednet.ucla.eduDisclosures: None

