

Expert Opinion / Conference Coverage · February 09, 2021
ASCO GU21: Abstract Recommendations (RCC)
by Robert A Figlin, MD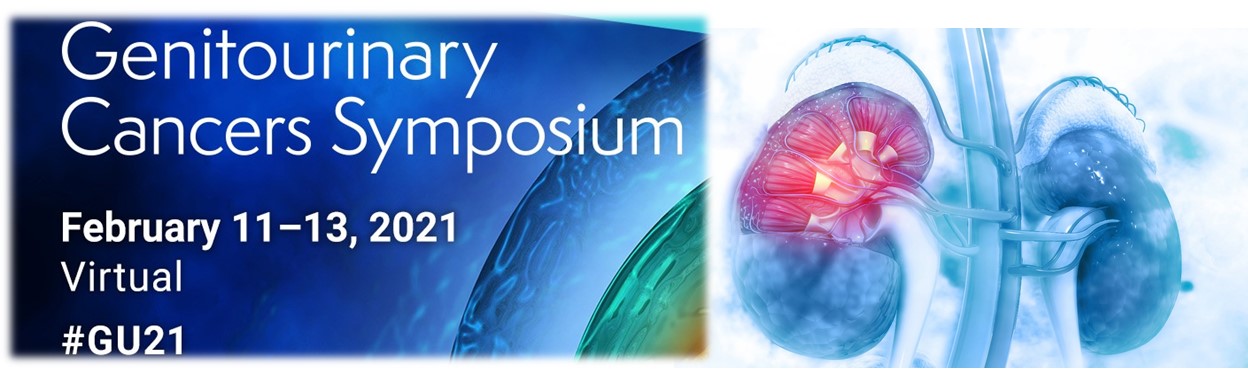

The abstracts included in this report have been selected by Robert A. Figlin, MD, Editor-in- Chief of the Kidney Cancer Journal. The chosen abstracts provided here highlight some of the most important trends in ongoing trials and as well as reflect the foremost research and strategies from latest clinical trials that impact the current standard of care in renal cancer.
ABSTRACT 269:

Background: In pts with advanced RCC, second-line treatment with LEN + EVE prolonged progression-free survival (PFS) compared with EVE alone. LEN + PEMBRO, also showed preliminary efficacious evidence in a phase 1/2 RCC study. Here, we describe the investigational study results of first-line LEN + PEMBRO or LEN + EVE versus SUN in pts with advanced RCC.
Methods: Pts were randomized (1:1:1) to receive LEN 20 mg orally once daily + PEMBRO 200 mg IV every 3 weeks (wks); or LEN 18 mg + EVE 5mg orally once daily; or SUN 50 mg orally once daily (4 wks on/ 2 wks off). Eligible pts had advanced RCC with no prior systemic therapy. Randomization was stratified by geographic region and MSKCC prognostic group. The primary endpoint was PFS by Independent Review Committee per RECIST v1.1. Secondary endpoints included overall survival (OS), objective response rate (ORR) and safety. A sequential approach was used to test PFS first, then OS and ORR. PFS and OS were compared across arms by a stratified log-rank test; hazard ratios (HRs) were estimated by a stratified Cox regression model.
Results:1069 pts were randomized (Table). After a median follow-up of 27 months (data cutoff August 28, 2020), PFS was significantly improved with LEN + PEMBRO (median 24 months [mos]) vs SUN (median 9 mos; HR 0.39, 95% CI 0.32–0.49) and LEN + EVE (median 15 mos) vs SUN (HR 0.65, 95% CI 0.53–0.80). OS was significantly longer with LEN + PEMBRO vs SUN (HR 0.66, 95% CI 0.49–0.88), whereas OS with LEN + EVE vs SUN was not statistically different (HR 1.15, 95% CI 0.88–1.50). ORR was significantly greater with LEN + PEMBRO (ORR 71%; complete response [CR] 16%) vs SUN (ORR 36%; CR 4%; odds ratio 4.35, 95% CI 3.16–5.97) and LEN + EVE (ORR 54%; CR 10%) vs SUN (odds ratio 2.15, 95% CI 1.57–2.93). Grade$3 treatment-related adverse events occurred in 72%of pts in the LEN + PEMBRO arm and 73% of pts in the LEN + EVE arm compared with59%of pts in the SUN arm. Conclusions: LEN + PEMBRO demonstrated significant improvements in PFS, OS and ORR vs SUN. LEN + EVE demonstrated significant improvements in PFS and ORR vs SUN. Safety was manageable and consistent with the known single-agent profiles.
Clinical trial information:NCT02811861.
Research Sponsor: Eisai Inc., Woodcliff Lake, NJ, USA; and Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA.
ABSTRACT 270:

Background: MET signaling is a key molecular driver in pRCC. Given that there is no optimal therapy for metastatic pRCC, we sought to compare an existing standard (sunitinib) to putative MET kinase inhibitors.
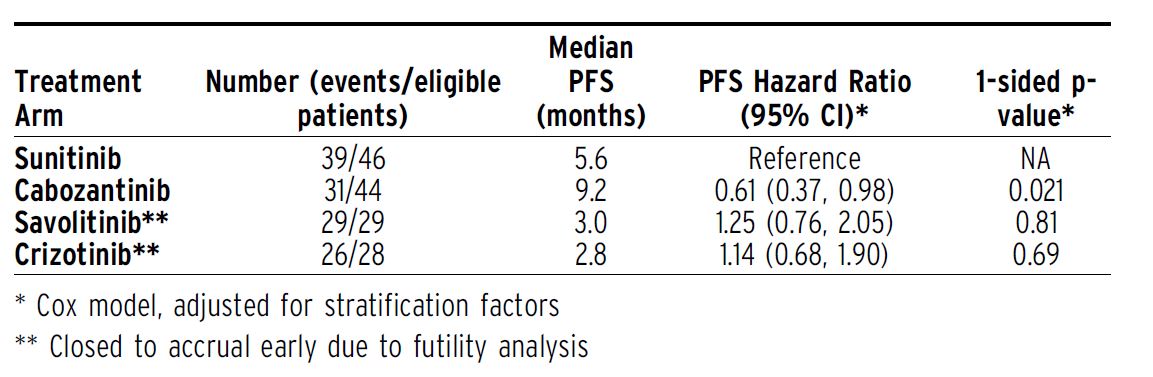
Methods: Eligible patients had pathologically verified pRCC, Zubrod performance status 0-1, and measurable metastatic disease. Patients may have received up to 1 prior systemic therapy excluding VEGF-directed agents. Patients were randomized 1:1:1:1 to receive either sunitinib 50 mg po qd (4 wks on/2 wks off), cabozantinib 60 mg po qd, crizotinib 250 mg po bid, or savolitinib 600 mg po qd. Patients were stratified by prior therapy and pRCC subtype (I vs II vs not otherwise specified [NOS]) based on local review. The primary objective was to compare progression-free survival (PFS) for each experimental arm versus sunitinib. With 41 eligible patients per arm, we estimated 85% power to detect a 75% improvement in median PFS with a 1-sided alpha of 0.10 using intent-to-treat analysis. A pre-planned futility analysis was performed after 50% of PFS events occurred. Secondary endpoints included toxicity, response rate, and overall survival.
Results:Between 4/2016 and 12/2019, 152 patients were enrolled; 5 were ineligible. Median age was 66 (range:29-89) and 76% were male; 92% had no prior therapy. By local pathologic review, 18%, 54% and 28% of patients were characterized as having type I, type II and NOS histology, respectively. In contrast, the frequency of type I, type II, and NOS by central review was 30%, 45% and 25%, respectively. Accrual to the savolitinib and crizotinib arms was halted early for futility (PFS hazard ratio . 1.0 for both); accrual continued to completion in the sunitinib and cabozantinib arms. Median PFS was significantly higher with cabozantinib relative to sunitinib (Table). Grade 3 or 4 adverse events occurred in 69%, 72%, 37% and 39% of patients receiving sunitinib, cabozantinib, crizotinib and savolitinib, respectively; one grade 5 adverse event was seen with cabozantinib. Overall survival and response data will be presented.
Conclusions: In this multi-arm randomized trial, only cabozantinib resulted in a statistically significant and clinically
meaningful prolongation of PFS in pRCC patients compared to sunitinib. These data support
cabozantinib as a reference standard for eligible patients with metastatic pRCC.
Clinical trial information: NCT02761057.
Research Sponsor: U.S. National Institutes of Health, Pharmaceutical/Biotech Company.ABSTRACT 272:

Background: Belzutifan (MK-6482) inhibits HIF-2a and demonstrated antitumor activity and favorable safety as monotherapy in a phase 1 study of patients (pts) with metastatic ccRCC. Current study (NCT03634540) investigates belzutifan plus cabozantinib for pts with advanced ccRCC who were either treatment naive (cohort 1) or previously treated, including immunotherapy and TKIs (cohort 2). This preliminary analysis presents data from cohort 2.
Methods: Pts had metastatic ccRCC and received no more than 2 prior systemic treatment regimens. Initially, 6 pts in either cohort 1 or 2 were treated with belzutifan 120 mg and cabozantinib 60 mg orally once daily for 21 days and a safety review committee performed an initial evaluation. For purpose of this preliminary analysis, efficacy was evaluated in pts who received $1 dose of treatment and had an opportunity of$6mo of follow-up. Primary end point: objective response rate (ORR; RECIST v1.1 by investigator review). Secondary end points: progression free survival (PFS), overall survival (OS), and duration of response (DOR). Safety was evaluated for all cohort participants.
Results:Evaluation of safety and tolerability of belzutifan 120 mg plus cabozantinib 60 mg was performed in the first 6 pts. Only 1 participant experienced a dose-limiting toxicity of hand-foot syndrome, therefore belzutifan 120 mg plus cabozantinib 60 mg was determined to be the recommended phase 2 dose. 53 pts were included in the safety analysis population. Median age was 64 yrs, 73.6% were male, 54.7% had ECOG PS 1. Twenty-eight (52.8%) received prior first-line and 24 (45.2%) prior second-line therapies. Median (range) time from enrollment to data cutoff was 11.3 mo (5.6- 24.0) for pts with $6 mo of follow-up (n=41). The confirmed ORR was 22.0% (9 PRs) and 90.2% had any tumor shrinkage. Disease control rate (CR+PR+SD) was 92.7%. Median (range) DOR was not reached (3.7+ to 14.8+ mo); all responses were ongoing. Median (95% CI) PFS was 16.8 mo (9.2- not reached); PFS rate at 6 mo was 78.3%. OS rate at 6 mo was 95.0%. While 52 of 53 (98.1%) pts experienced a treatment-related adverse event (TRAE), 92% of events were grade 1 and 2. Most common ($30%) TRAEs were anemia (75.5%), fatigue (67.9%), hand-foot syndrome (52.8%), diarrhea (45.3%), hypertension (43.4%), nausea (35.8%), and ALT/AST increase (32-34%). Incidence of grade 3 TRAEs .5% were hypertension (22.4%), anemia (11.3%), fatigue (11.3%), and ALT increase (5.7%). 2 pts experienced grade 3 hypoxia (3.8%). There were no grade 4 TRAEs or deaths. Discontinuations due to TRAEs occurred in 6 pts (11.3%) for belzutifan and 8 pts (15.1%) for cabozantinib.
Conclusions: In this preliminary analysis, belzutifan in combination with cabozantinib demonstrated promising antitumor activity in previously treated pts with metastatic ccRCC. Safety was consistent with individual profiles of each agent. Clinical trial information: NCT03634540. Research Sponsor: Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA.
ABSTRACT 285:

Background: In the phase III, open-label CheckMate 9ER trial (NCT03141177), patients with aRCC were randomized 1:1 (stratified by International Metastatic Renal Cell Carcinoma Database Consortium risk score, tumor programmed death ligand 1 expression, geographic region) to nivolumab 240 mg IV Q2W + cabozantinib 40 mg PO QD (N+C; n = 323) or sunitinib (S) 50 mg PO (4 weeks of 6- week cycles; n = 328) for first-line treatment until disease progression or unacceptable toxicity (max N treatment, 2 years). N+C met primary and secondary efficacy endpoints by significantly improving progression-free survival, overall survival, and objective response rate versus S in aRCC patients with a clear cell component. Here, we present in-depth health-related quality of life (HRQoL) patient-reported outcome (PRO) results, including overall between-group comparisons of treatment groups and time to confirmed deterioration (TCD).
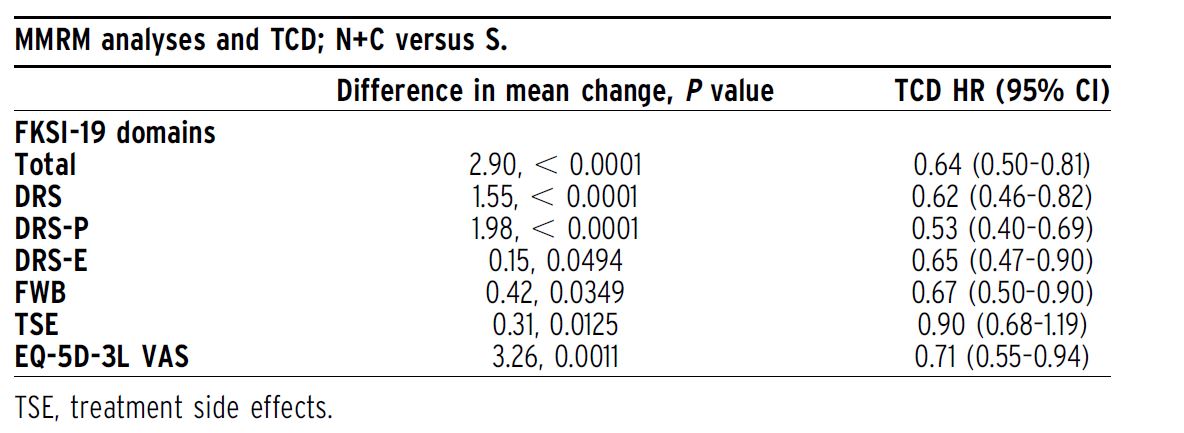
Methods: PROs in all randomized patients were an exploratory endpoint assessed using the Functional Assessment of Cancer Therapy Kidney Symptom Index-19 (FKSI-19) and EQ-5D-3L instruments. PRO assessments at baseline, common on-treatment scheduled visits, and common follow-up visits for both arms were analyzed. Changes from baseline were assessed using mixed-model repeated measures (MMRM), adjusting for baseline scores and stratification factors. TCD was calculated from Kaplan–Meier estimates and Cox proportional hazards models.
Results:Median follow-up for overall survival was 18.1 months. PRO completion rates were . 90% at baseline, and $ 80% at all on-treatment assessments ($ 10 patients) through week 91 in both arms. The overall least squares mean difference in change from baseline favored N+C over S in FKSI-19 (all domains) and in EQ-5D-3L. Patients treated with N+C experienced less treatment burden, with decreased risk of confirmed deterioration across most measurements versus S, including FKSI-19 total, disease-related symptoms (DRS), DRS-physical (DRS-P), DRS-emotional (DRS-E), functional well-being (FWB), and EQ- 5D-3L visual analog scale (VAS) scores (Table).
Conclusions: Patients reported statistically significant HRQoL benefits with N+C versus S. Treatment with N+C significantly reduced the risk of deterioration in HRQoL scores, including in disease-related symptoms of kidney cancer. These results suggest that the superior efficacy of N+C over S comes with the additional benefit of improved HRQoL. Clinical trial information: NCT03141177. Research Sponsor: Bristol Myers Squibb.
ABSTRACT 308:

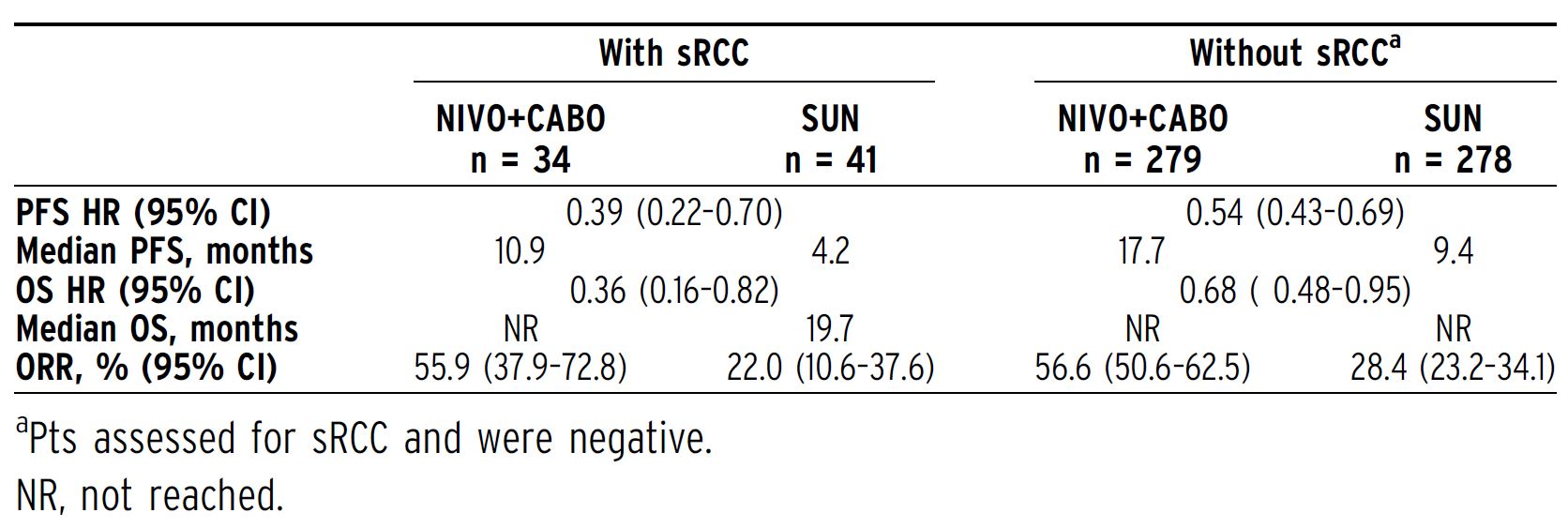
Background: First-line NIVO+CABO met primary and secondary efficacy endpoints by improving progression-free survival (PFS; HR 0.51, P,0.0001), overall survival (OS; HR 0.60, P = 0.0010), and objective response rate (ORR; 55.7% vs 27.1%; P , 0.0001) vs SUN in patients (pts) with aRCC in CheckMate 9ER (Choueiri et al. ESMO 2020). Efficacy benefits with NIVO+CABO vs SUN were consistent across prespecified subgroups including by IMDC risk group, and regardless of tumor PD-L1 expression (database lock for primary analysis, March 30, 2020). Updated analyses are needed to establish durability of benefit with first-line NIVO+CABO and assess outcomes in aRCC pts with sarcomatoid features (sRCC)—an aggressive histologic subtype associated with poor prognoses.
Methods: In this phase III open-label trial, adults with confirmed aRCC (with a clear cell component including those with sRCC) were randomized 1:1 (stratified by IMDC risk score, tumor PD-L1 expression, geographic region) to NIVO 240 mg IV Q2W + CABO 40mg PO QD vs SUN 50 mg PO (4 weeks of 6-week cycles). The primary endpoint was RECIST v1.1-defined PFS by blinded independent central review (BICR) in all randomized (intent-to-treat [ITT]) pts; secondary endpoints included OS, ORR by BICR, and safety. Pts with and without sRCC were identified by local pathology report, and outcomes in these pts were evaluated via prespecified supportive subset analyses.
Results:The presence of sRCC was assessed in ITT pts (N = 651) at enrollment. Overall, 75 (11.5%) pts had sRCC and 557 (85.6%) did not; sRCC status was not reported in 19 pts (2.9%). Overall, 34 vs 41 pts with sRCC were randomized to NIVO+CABO vs SUN, respectively. At a median follow-up of 18.1 months, NIVO+CABO improved PFS, OS, and ORR in sRCC pts vs SUN (Table). Notable PFS, OS, and ORR benefits were observed with NIVO+CABO vs SUN in the subgroup of pts without sRCC. Median PFS was doubled, the risk of death was lower, and ORR was consistently higher with NIVO+CABO vs SUN regardless of sarcomatoid status. Key updated PFS, OS, response, and safety outcomes in the ITT population and in pts with and without sRCC will be reported with additional follow-up based on a September 10, 2020 database lock.
Conclusions: NIVO+CABO demonstrated improved efficacy and prolonged survival vs SUN in previously untreated aRCC pts regardless of sarcomatoid status. Updated results with extended follow-up will assess the durability of outcomes in this trial. Clinical trial information: NCT03141177. Research Sponsor: Bristol Myers Squibb.
ABSTRACT 309:

Background: The long-term efficacy and tolerability of nivolumab (NIVO) 3 mg/kg + ipilimumab (IPI) 1 mg/kg Q3W 3 4 doses followed by NIVO 3 mg/kg Q2W for previously untreated advanced RCC (aRCC) demonstrated in the registrational CheckMate 214 clinical trial was based on patients (pts) with a predominantly clear cell component. CheckMate 920 (NCT02982954) is a US community-based, multi-arm, phase IIIb/IV clinical trial of NIVO+IPI treatment in pts with previously untreated aRCC and clinical features mostly excluded from phase III trials. Here, we present the safety and efficacy results for the cohort of pts with nccRCC from CheckMate 920, a patient population with a poor prognosis and without a definitive effective treatment.
Methods: Pts with previously untreated advanced/metastatic nccRCC, Karnofsky performance status $ 70%, and any International Metastatic Renal Cell Database Consortium risk received NIVO 3 mg/kg + IPI 1 mg/kg (NIVO3+IPI1) Q3W 3 4 doses followed by NIVO 480 mg Q4W for # 2 years or until disease progression/unacceptable toxicity. The primary endpoint was incidence of any-causality grade$3 immune-mediated adverse events (imAEs) within 100 days of last dose of study drug. Key secondary endpoints: progression-free survival (PFS) and objective response rate (ORR) by RECIST v1.1 (both per investigator), duration of response (DOR), and time to response (TTR). Exploratory endpoints included overall survival (OS).
Results:Of 52 treated pts with nccRCC, 69.2% were men; median age was 64 years (range, 23–86), and 28.8% had sarcomatoid features. Histological subtypes were papillary (34.6%), chromophobe (13.5%), translocation associated (3.8%), collecting duct (3.8%), renal medullary (1.9%), or unclassified (42.3%). With 24.1 months minimum follow-up, median duration of therapy (range) was 3.5 months (0.0–25.8) for NIVO and 2.1 months (0.0–3.9) for IPI. Median (range) number of doses received was 4.5 (1–28) for NIVO and 4.0 (1–4) for IPI. No grade 5 imAEs occurred. Grade 3–4 imAEs (n = 52) by category were diarrhea/ colitis (7.7%), rash (5.8%), nephritis and renal dysfunction (3.8%), hepatitis (1.9%), adrenal insufficiency (1.9%), and hypophysitis (1.9%). ORR (n = 46) was 19.6% (95% CI, 9.4–33.9). Two pts achieved complete response (papillary, n = 1; unclassified pathology, n = 1), 7 achieved partial response (papillary, n = 4; unclassified pathology, n = 3), and 17 pts had stable disease. Median TTR was 2.8 months (range, 2.1–4.8). Median DOR was not reached (range, 0.03+–27.8+); 8 of 9 responders remain without reported progression. Median PFS (n = 52) was 3.7 months (95% CI, 2.7–4.6). Median OS (n = 52) was 21.2 months (95% CI, 16.6–not reached).
Conclusions: In pts with previously untreated nccRCC, a population with high unmet medical need, treatment with NIVO3+IPI1 Q3W followed by NIVO 480 mg Q4W showed no new safety signals, and encouraging antitumor activity. Clinical trial information: NCT02982954. Research Sponsor: Bristol Myers Squibb.©
ABSTRACT 313:

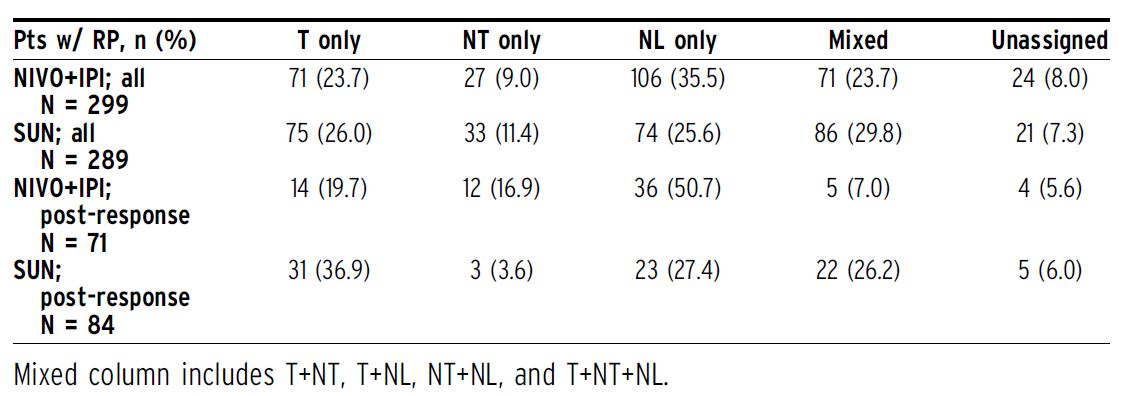
Background: First-line NIVO+IPI demonstrates superior survival and response benefits in intentto- treat (ITT) patients (pts) with aRCC after long-term follow-up in the phase 3 CheckMate 214 trial. Data are scarce on tumor relapse and patterns of disease progression with immuno-oncology agents in this setting. This exploratory analysis of CheckMate 214 characterizes patterns of progression with NIVO+IPI vs SUN with 4 years minimum follow-up.
Methods: Pts with clear cell aRCC were randomized to NIVO+IPI Q3W34 followed by NIVO monotherapy Q2W, or SUN QD34 weeks (6-week cycle). Patterns of progression were characterized in ITT pts and analyzed post hoc using descriptive statistics. Progression patterns were defined by $ 20% target lesion growth (T), unequivocal progression of nontarget lesions (NT), and new lesion(s) (NL). Response and progression were assessed per independent radiology review committee via RECIST v1.1.
Results:Radiographic progression (RP) was documented in 299/550 (54.4%) ITT pts with NIVO+IPI vs 289/546 (52.9%) with SUN. Among ITT pts with a confirmed response (objective response = 215/550 [39.1%, NIVO+IPI] vs 177/546 [32.4%, SUN]), 71/215 (33.0%) vs 84/177 (47.5%) pts experienced post-response RP with NIVO+IPI vs SUN; 8/59 (13.6%) vs 3/14 (21.4%) progressed after complete response, and 63/156 (40.4%) vs 81/163 (49.7%) progressed after partial response, respectively. The pattern of RP differed between arms (Table). With NIVO+IPI, 106/299 (35.5%) RPs resulted from NL only vs 74/289 (25.6%) with SUN, and this differential was more pronounced in pts with an initial confirmed response (36/71 [50.7%] vs 23/84 [27.4%]). Most NL-only RPs in initial responders occurred in a single organ (34/36 [94.4%] for NIVO+IPI; 20/23 [87.0%] for SUN) with the most common being lymph nodes (11/34 [32.4%]), brain (8/34 [23.5%]), and lung (5/34 [14.7%]) with NIVO+IPI, and lymph nodes (7/20 [35.0%]), brain (4/20 [20.0%]) and adrenal gland (3/20 [15.0%]) with SUN. Additional progression details, baseline characteristics, and key efficacy outcomes in progressors will be reported.
Conclusions: Differential patterns of tumor relapse and disease progression were observed after long-term follow up of patients treated with NIVO+IPI vs SUN in CheckMate 214. NL-only progression occurred more often with NIVO+IPI vs SUN, in particular in the subset of pts who progressed post-response. These patterns may have therapeutic implications. Clinical trial information: NCT02231749. Research Sponsor: Bristol Myers Squibb.
ABSTRACT 333:

Background: Patients (pts) with VHL disease are at risk of developing benign and malignant tumors, including ccRCC, pancreatic lesions tumors, CNS hemangioblastomas, and retinal lesions. Inactivation of VHL leads to stabilization of HIF-2a, which drives tumor growth. In a phase 1/2 study, MK-6482, a potent, selective, oral small molecule HIF-2a inhibitor, demonstrated favorable safety and antitumor activity in advanced ccRCC. We present results of the open-label phase 2 study of MK-6482 for VHL disease–associated ccRCC (NCT03401788).
Methods: Adults with germline VHL alterations, measurable, localized/non-metastatic ccRCC, no prior systemic anticancer therapy, and ECOG PS 0/1 received MK-6482 120 mg once daily until progression, intolerable toxicity, or decision to withdraw. Primary end point: ORR of VHL-associated ccRCC tumors per RECIST v1.1 by independent review committee (IRC). Secondary end points: DOR, time to response (TTR), PFS, and safety.
Results:As of June 1, 2020, 61 pts enrolled. The majority (82%) of pts had ECOG PS 0, and median number of prior surgeries per pt was 5 (range, 1-15). Lesions outside the kidney (non-RCC tumors) evaluable by IRC included pancreatic lesions (100%) and CNS hemangioblastomas (70%). Median duration of treatment was 68 wk (range, 8-105), and 92% of pts remain on therapy. There were 22 confirmed responses (ORR, 36% [95% CI, 24%-49%]) and 7 (11%) unconfirmed (documented at 1 time point, to be confirmed at subsequent time point) responses; all PRs. In pts with confirmed PR, median DOR was not reached (range, 12-62 wk) and median TTR was 31 wk (range, 12- 61); 14 (64%) pts had response duration of$26 wk. 56 pts (92%) had any decrease in size of target lesions. PFS rate at 52 wk was 98% (95% CI, 89%-100%). Overall, pretreatment median linear growth rate of ccRCC tumors was +3.6 mm/y (range, 23.4 to +33.1), compared with 24.5 mm/y (range,212.8 to +5.1) while on treatment. For non-RCC tumors, ORR in pancreatic lesions was 64% (39/61, including 4 CRs) and in CNS hemangioblastomas was 30% (13/43, including 5 CRs). Median (range) TTR was 35 wk (11-60) and 12 wk (10-60) for pancreatic lesions and CNS hemangioblastomas, respectively. 11/16 (69%) pts with evaluable retinal lesions at baseline showed improvement. Of those 16 pts, 29 eyes were followed for retinal lesions; 16 eyes (55%) showed improvement, 12 (41%) remained stable, and no follow-up evaluation was available for 1 (3%) eye. Treatment-related AEs (TRAEs) occurred in 98% of pts, none grade 4/5. Most common TRAE was anemia (87%), considered to be an on-target toxicity. One pt discontinued treatment due to a TRAE (grade 1 dizziness). As of data cutoff, 1 pt (2%) required surgery for ccRCC tumors after treatment.
Conclusions: MK-6482 is an active and well-tolerated therapy for VHL disease– associated ccRCC, pancreatic lesions, as well as CNS and retinal hemangioblastomas. Clinical trial information: NCT03401788. Research Sponsor: Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA.
What To Expect from ASCO GU21 - GUIDE
