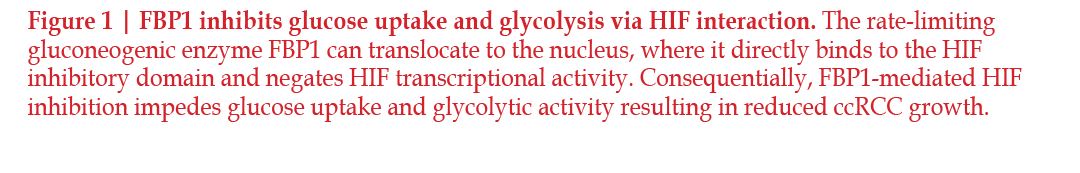It’s Clear as Day: HIF Signaling is Driving Force of the Clear Cell Morphology
Whitney A. Brown, W. Kimryn Rathmell*, Zachary A. Bacigalupa*
Department of Medicine, Vanderbilt University Medical Center, Nashville, TN 37232
ABSTRACT
Clear cell renal carcinoma (ccRCC) is the most common form of kidney cancer
with few therapeutic options in its advanced stages. ccRCC has genetic
predisposition linked to the von Hippel Lindau gene. The product of this gene
is responsible for proteasomal degradation of the hypoxia induced factors, which
when stabilized activate hundreds of pathways, some of which promote tumor growth
via angiogenesis, and upregulating glycogen and lipid biosynthesis. The “clear cell”
morphology exhibits a large, translucent cytoplasm attributed to excessive glycogen
and lipid deposition. Biochemical analyses have demonstrated that these lipid
depots in ccRCC are enriched with high concentrations of high-density lipoprotein
cholesterol, which is known to play an integral role in membrane rigidity and drug
resistance. Glycogen synthesis serves as an energy source for tumoral growth, and lipid
and cholesterol buildup within tumors has been linked to the formation of new cell
membranes for cellular growth. In this review we will summarize how glycogen, lipid,
and cholesterol metabolism play key roles in ccRCC tumor growth and the therapeutic
potential of targeting these pathways.
INTRODUCTION
Clear cell renal cell carcinoma
(ccRCC) is the most common
form of kidney cancer, accounting
for 70-75% of all kidney cancers,
which affects males twice as often as females
¹. Current therapies include tyrosine
kinase inhibitors (TKI) targeting
factors involved in angiogenesis, which
is essential for ccRCC tumor growth²,
³, immunotherapies, targeting checkpoints
regulating T cell activation4, and
the combination of both5. Identifying
strategies to enhance the efficacy of current
therapeutics, or to achieve durable
disease control with reduced toxicity,
has become the focus of current investigations.
ccRCC is linked to genetic
factors that control cell metabolism,
which makes it a ripe target for studying
the oncologic metabolic shift known as
the Warburg effect5 as a potential therapeutic
angle. The Warburg effect describes
a dependence on aerobic glycolysis
and lactic acid fermentation, while
the tricarboxylic acid (TCA) cycle is
downregulated even in the presence of
oxygen. Studies have shown an increase
in glucose uptake and aerobic glycolysis
6-9. Fewer TCA intermediates were
present in ccRCC, further confirming a
shift towards aerobic glycolysis and indicating
that pyruvate dehydrogenase
is less active in ccRCC6,10. This discovery
also demonstrates that ATP production
is dependent on aerobic glycolysis
rather than oxidative phosphorylation6,10. Within the TCA cycle, fumarate and
malate levels were lower than normal
tissues, while succinate, isocitrate, and
citrate were higher, indicating a dependence
on reductive carboxylation
through citrate8,9. This upregulation of
reductive carboxylation was shown to
be the route for fatty acid synthesis in
ccRCC11-13. Given that a Warburg shift
is a complex matter with many intermediates,
this discovery in ccRCC provides
multiple targets for therapeutic
interventions; currently glutaminase
inhibitors are being examined as target
to prevent the formation of citrate, and
therefore prevent reductive carboxylation
in ccRCC13.
These genetic predispositions
in ccRCC are linked to chromosome 3
translocations, deletions, and mutations
that effect the von Hippel Lindau
(VHL) gene and its expression. This
molecule is well known as a major effector
of the hypoxia response, as the
key negative regulator of the hypoxia
inducible factors (HIF), a potent family
of transcription factors and their downstream
transcriptional targets such
as vascular endothelial growth factor
(VEGF)14–16. HIFs interact with the
product of VHL (pVHL) through oxygen
dependent domains that are targeted
prolylhydroxylation enzymes
15,17–19. Under normal oxygen conditions,
pVHL forms a ubiquitin ligase complex
that recognizes hydroxylated proline
residues and binds to the alpha subunit
of HIF, leading to its polyubiquitination
and degradation16. In hypoxic conditions
HIF-α is not recognized by pVHL,
allowing it to dimerize with HIF-ß. This
dimer is an essential transcriptional
regulator of hundreds of genes and signaling
cascades that promote hypoxic
adaptation16, such as the activation of
vascular endothelial growth factor receptor
(VEGFR) signaling20. The HIF transcriptional network activates many
enzymes and proteins integral to key
metabolic pathways whose enhanced
activity promotes tumor growth when
pVHL is absent21, 22.
The alpha subunit of HIF is
present in two main forms—HIF-1α
and HIF-2α. These both have different
functions in the cell and presentation in
ccRCC, and this distinction is critical for
discussions of metabolism. Although
both HIF factors are targets of pVHL,
HIF-1α is not always present in ccRCC,
and VHL-mutated tumors can be classified
as expressing both HIF-1 and HIF-2
(HIH2), or HIF-2 only (H2)23. The downregulation
of HIF-1α is one feature that
drives more aggressive disease states16
and suggests that HIF-1α has tumor
suppressor functionality in ccRCC.
While HIF-1α expression and activity
cannot completely counteract the oncogenic
effects of HIF-2α, its presence
can decrease the severity of the prognosis16.
When stabilized, HIF-1α, as a
transcription factor, has potent effects
on genes involved in activating aerobic
glycolysis24,25. HIF-2α is expressed in
all VHL-/- ccRCC and its elimination
in these cells prevents tumor growth.
The role of HIF-2α inhibition is to block
HIF-2α transcription and therefore inhibit
its downstream targets, such as
VEGF, as well26. Studies have shown decreased
tumor formation in xenograft
models when HIF-2α is inhibited and
pVHL is absent27-29. An effective mechanism
of inhibition has been identified
as inhibiting translation of HIF-2α by
targeting the binding of its iron responsive
element (IRE)27, 30–32. This study
showed that hypoxia increases HIF concentration
via a 5’-UTR IRE that binds
to iron responsive protein 1 (IRP1), and
when exogenous iron is added, translation
of HIF proteins increases30, 33*.
Additionally, a recent study showed via
proximity ligation assays that an inhibitor
of HIF-2α, PT2385, decreased HIF-2α
complexes in ccRCC biopsies analyzed
before and during treatment3434. In this
study, they measured efficacy based on
three factors: (1) the concentration of a
downstream target of HIF-2α, erythropoietin
(EPO), (2) the dissociation of
HIF-2 complexes, and (3) the amount
of gene expression. They found significantly
decreased levels of EPO in 90% of
patients after two weeks, showing the
HIF inhibition was effective34. Using
fluorescently conjugated antibodies
for HIF-2α and HIF-1β, they were able
to detect proximity via florescence microscopy
to show a significant decrease
in HIF-2α complexes during drug treatment
as compared to pretreatment observations
in two of three patient samples,
and via RNA-seq analysis they
found that 277 genes were downregulated
by the inhibitor in those same
two patients34. Complex dissociation
and gene expression were found to be
correlated to one another, indicating
that downregulation of HIF-2α dependent
genes may be necessary for antitumor
activity34. Since this inhibitor
was shown to have high variability, it
was later improved to PT2977 and is
now known as MK6482. The improvements
were made with the goal of improving
pharmacokinetic stability by
decreasing binding to serum proteins,
increasing the binding affinity for the
HIF-2α binding pocket, and lowering
the susceptibility of glucuronidation to
a key hydroxyl group26, 35–37. A phase I
trial with MK6482 concluded that 67%
of patients had reduced target-lesion
size with manageable anemia being the
most common adverse event, and hypoxia
being the only adverse event that
caused patient discontinuation/dosage
reduction26, 38, 39. A phase II trial used a
cohort of patients with VHL-associated,
nonmetastatic ccRCC; 87% of the cohort
had decreased tumor size26, 40. A phase
III trial is currently being conducted to
compare the efficacy of MK6482 versus
everolimus26, 41. The mechanism
of resistance to HIF-2α inhibitors has
been identified as either mutations
that prevent drug binding or mutations
that increase HIF stabilization26,
34, 42, but newer HIF-2α inhibitors have
the potential to overcome these mutation
barriers by using a combinatorial
approach, targeting factors that are implicated
when resistance occurs26, 43–47.
Inhibitors of HIF-2α show great clinical
promise alongside other targets in
ccRCC.
Another target with approved
therapies for RCC treatment
is the mammalian target of rapamycin
(mTOR). This classical metabolism
regulator is a serine/threonine kinase
that functions as a nutrient sensor by
responding to environmental conditions,
such as changes to oxygen levels,
metabolite abundance, amino acids and
growth factors48. Rapamycin (sirolimus),
and rapamycin analogs everolimus
and temsirolimus, block mTOR
activity by forming a gain-of-function
complex with FK506-binding-protein
(FKBP12)12–14. This complex acts as an
allosteric inhibitor of mTOR complex
1 to accomplish this inhibitory effect48,
51. In addition to regulating metabolic
responses, this factor acts upstream of
VEGFR to further promote angiogenesis.
In vitro experiments have shown
that inhibition of mTOR prevents angiogenesis
and tumor growth as well as
decreasing lipogenesis48. We will continue
to discuss specific targets within
glycogen metabolism, lipid metabolism,
and cholesterol metabolism for the
remainder of this review.
Glycogen Metabolism
ccRCC is classified by highly regulated
lipid and glycogen metabolisms and increased
deposits in the cell for both52.
In general, activation of glycolysis and
inactivation of the TCA cycle is associated
with ccRCC and explains the energy
supply for the tumor53. Furthermore,
there is evidence that oxidative phosphorylation
is inhibited in ccRCC,
which further supports that the energy
supply of these tumors is dependent on
glycolysis53. Specifically, high concentrations
of glycolytic enzymes, which
are supported by a hypoxic microenvironment,
and low concentrations of
TCA cycle intermediates are found in
these tumor cells52. In ccRCC cells, lactate
is also upregulated, in part due to
transcriptional activation of Lactate
Dehydrogenase (LDH), further suggesting
that the cells function on aerobic
glycolysis52, 54.
Although these trends are seen
across the spectrum of ccRCC tumors,
quantitatively, glycogen and lipid deposits
are tumor grade dependent, with
glycogen and lipid accumulation more
prevalent in lower grade tumors54.
These features have been linked to
prognostic algorithms, such as the transcriptional
ccA and ccB signature55, 56.
Further investigations into the metabolic
shifts associated with stage progression
are being described with increasing
frequency, most recently with the
Cancer Genome Atlas index paper on
ccRCC5, 7 and dedicated metabolomic
profiling9. Finally, failure of antitumor
therapies has also been linked to the
expression of glycolytic and hypoxia
factors and presumed upregulation of
compensatory signaling pathways52*.
Glycolysis and glycogen synthesis
are regulated by several factors in
the cell. As discussed previously, mTOR
promotes tumor growth and angiogenesis
in ccRCC. One way mTOR accomplishes
this is by activating glycolysis
and glycogen synthesis, providing an
energy source for the tumors. A recent
study showed that the phosphoinositide
3-kinase (PI3K)-protein kinase B (AKT)-
mTOR signaling axis is associated with
the progression of ccRCC57. Human
ccRCC cell lines CAKI-1 and RCC4 were
treated with NVP/MEZ235, a dual inhibitor
of both PI3K and mTOR, and
showed decreased phosphorylation of
AKT protein and mTOR. By effectively
blocking AKT and mTOR activation,
the researchers observed significant inhibition
of glycolysis and glycogen synthesis,
removing the energy source and
decreasing tumoral growth57. As a tyrosine
kinase that orchestrates a robust
signaling cascade regulating many biosynthetic
processes, PI3K has long been
an integral target for TKI treatments58.
Another key regulator of glucose
metabolism is glycogen synthase 1
(GYS1)59. Glycogen synthase is a major
regulator of glycogen catabolism which,
when active, promotes the synthesis of
glycogen. A recent study showed that
GYS1 is significantly overexpressed in
ccRCC tumors and was mostly found
in the cytoplasm, which is where glycogen
synthesis occurs. This overexpression
was then correlated to poor
overall survival in the clinical setting59.
Additionally, this study showed in a
western blot that p65 expression increased
when GYS1 was overexpressed
via, indicating that GYS1 interacts with
the canonical NF-κB pathway. Glycogen
synthase is inactivated in the body by
glucagon and epinephrine, so finding
treatments that mimic these effects in
tumor cells and treating in combination
with inhibitors of glycolysis, could be
an area for further investigation.
In addition to factors that promote
the expression and activity of
glycolytic enzymes for energy generation,
several cellular modifications
have been observed which suggest the
regulation of this bioenergetic pathway
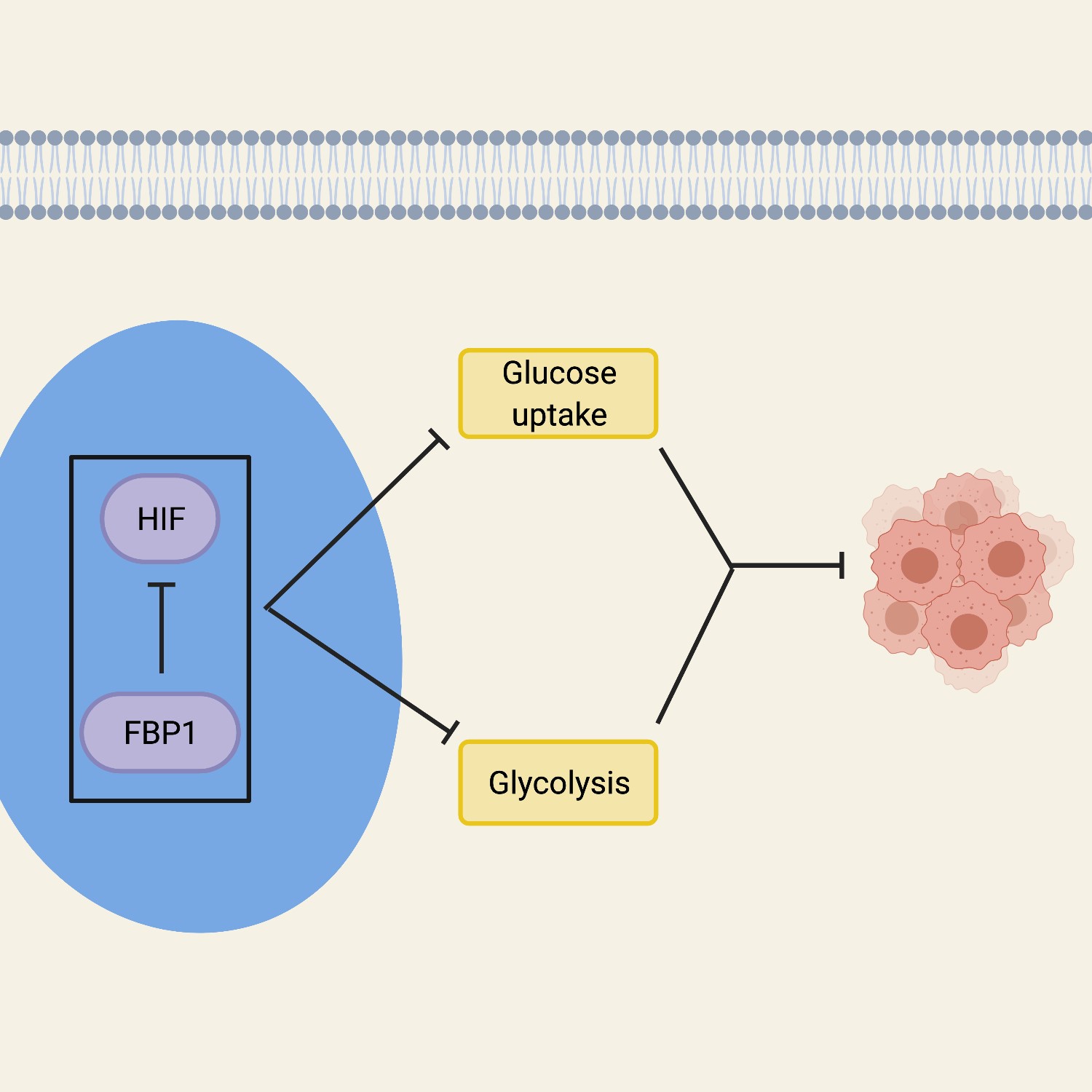
is tightly controlled. Fructose-1,6-
bisphosphatase 1 (FBP1) is a rate-limiting
gluconeogenic enzyme that plays
a large role in glucose metabolism and
inhibits HIF proteins in the nucleus54,
60. FBP1 opposes ccRCC by inhibiting
glycolysis and cell proliferation in
cells52, 60. Inhibition of FBP1 increases
glucose uptake and, therefore, allows
tumor growth to progress. Evidence
supported by cellular fractionation and
immunofluorescent staining suggests
that FBP1 suppresses HIF proteins in
the nucleus, and showed that an interaction
between FBP1 and HIF proteins
is necessary for an effect on glucose metabolism60.
This was further proven by
using a nuclear-excluded form of FBP1
which failed to inhibit the HIF proteins
in the cell, showing that the effects of
FBP1 inhibition originate in the nucleus60.
Overall, the FBP1 activity in the
cell that affects the growth and development
of tumors, works by regulating
HIF from the nucleus. The inhibition
of FBP1 promotes glycolytic functions,
thereby enhancing the Warburg effect,
while simultaneously failing to suppress
nuclear HIF function, both of
which is associated with poor prognosis
in ccRCC (Figure 1).
Lipid Metabolism
In ccRCC, lipid metabolism is an important
factor for tumor cell growth
because it provides the membrane
structures for the newly formed tumor
cells. Specifically, lipid droplet buildup
serves as fuel for membrane synthesis
for these tumor cells24–26. This process
of lipid droplet buildup occurs through
increased lipogenesis via reductive carboxylation
in parallel with the inhibition
of beta-oxidation11–13, 61. Evidence
shows that increased lipid storage
in ccRCC cells is associated with increased
tumorigenesis, and there is a
correlation between lipid metabolism
and ccRCC risk score62, 63. A recent
study looked into the effects of VHL
status on lipid catabolism versus lipid
uptake. By staining with Oil red O to
assess changes to the presence of lipid
droplets, Du et al. observed a decrease
in lipid droplets in cells where VHL
was reconstituted, suggesting that the
presence of pVHL impacts either lipid
uptake/synthesis or promotes lipid catabolism62.
In an effort to interrogate
the effect on lipid uptake, this study
tracked the uptake of BODIPY fluorescent
fatty acid dyes and concluded that
lipid uptake occurred independently
from VHL status62. Therefore, lipid
deposition is VHL-mediated while lipid
uptake occurs independently of VHL,
indicating that de novo lipid synthesis
is the major contributor to lipid droplet
formation in VHL-/- ccRCC62. Several
factors in the cell regulate this process
and are currently being studied as
points of therapeutic intervention.
One regulator of interest is
Kruppel life factor 6 (KLF6). KLF6 is a
zinc finger family transcription factor
that was shown to have effects on lipid
metabolism64 and has been implicated
as a tumor promoting factor in ccRCC
via its effects on cell proliferation and
high levels of expression. The gene encoding
this transcription factor was
found to be located within a locus containing
one of the strongest super enhancers.
Additionally, this association
was linked to enhanced KLF6 expression
when comparing ccRCC samples
to adjacent normal tissue, as well as to
other solid tumors lacking this super
enhancer. The Cancer Genome Atlas
data of ccRCC showed a correlation
between HIF-2α expression and KLF6
expression; this study investigated this
interaction through VHL reintroduction
experiments64. The reintroduction
of VHL caused a decrease in mRNA
expression of KLF6 and, using ChIPseq,
they showed that VHL introduction
caused a decrease in activity in
the region where the super enhancer is
located64. Additionally, the ChIP-seq
data show that HIF-2α was bound at
this same region64. This indicates that
HIF-2α is an activator of this super enhancer,
so when HIF-2α is present, it
binds to the super enhancer and there is
robust transcription of KLF6. To expand
on their findings, the researchers next
assessed the impact of altering KLF6
expression in ccRCC. Pathway analysis
was performed on RNA-seq data collected
from cells depleted of KLF6 and
revealed a significant downregulation
of lipid and cholesterol metabolism
pathways64. Specifically, they identified
sterol regulatory element binding
protein 1 and 2 (SREBP1 and SREBP2),
master transcriptional regulators of
lipid signaling, were downregulated in
response to KLF6 suppression. These
findings were validated with qPCR experiments,
where it was observed that
SREBP1, SREBP2, and several of their
downstream targets were downregulated
in response to KLF6 inhibition.
Importantly, these results translated
further into an overall decrease in intracellular
cholesterol and lipids when
KLF6 is depleted. These studies elegantly
display the critical role HIF-2α plays
in regulating KLF6, an essential piece
of lipid and cholesterol metabolism in
ccRCC.
mTOR signaling through
mTORC1 also regulates SREBP1 and
SREBP2. Investigations into the interaction
between mTORC1 and KLF6 revealed
that KLF6 both directly interacts
with SREBP1 and SREBP2, and promotes
mTOR signaling by enhancing
platelet-derived growth factor subunit
B (PDGFB); both of these factors contribute
to an increase in lipid metabolism
and anabolic signaling, resulting
in increased tumor growth64 (Figure 2).
SREBP acts by inducing the production
of enzymes involved in cholesterol and
lipid synthesis, including the rate-limiting
enzyme of cholesterol synthesis,
3-hydroxy-3-methyl-glutaryl-coenzyme
A reductase (HMGCR)65–67.
A recent study showed that the gene
TRC8 represses the translation of these
key transcription factors, therefore inhibiting
lipid and cholesterol synthesis,
which makes it a target for future
investigation65.
HIF proteins promote lipid metabolism
via a variety of mechanisms.
HIF proteins promote dietary lipid
uptake, interact with the gene PLIN2
to promote lipid storage, and interacts
the gene encoding carnitine palmitoyl
transferase 1 (CPT1A) to promote
lipid droplet formation. Lipid droplet
formation was shown to be HIF protein
dependent; cells that were double
knockdown for HIF-1α and HIF-2α had
a significant decrease in lipid droplet
formation62. Additionally, this study
showed that HIF-1α and HIF-2α bind
specifically to a CPT1A promoter via
ChIP analysis with HIF-1α and HIF-
2α antibodies in 12 regions identified
as HIF response elements62. A recent
study showed that dietary lipid uptake
leading to increased lipid in the kidneys
being driven by HIF-1α signaling
in human ccRCC12. The gene PLIN2 was
found to be over expressed in ccRCC
and suggests an interaction with HIF-
2α allows for heightened lipid storage.
The mechanism by which this occurs is
through stabilization of the endoplasmic
reticulum (ER). The interaction between
PLIN2 and HIF-2α is required to
maintain ER homeostasis and prevents
cell death under stressful conditions68.
This is a possible explanation for drug
resistance; when the ER is targeted by
therapeutic interventions, this interaction
could be preventing apoptosis.
Another study further analyze the HIF
dependence of lipid droplet formation
by focusing on the interaction between
HIF proteins and the gene encoding
CPT1A, which is a major regulator of
lipid synthesis. When CPT1A was in
low concentrations, it has shown increased
lipid storage associated with
tumorigenesis. It was discovered that
HIF-1α and HIF-2α directly bind with
CPT1A to inhibit its function and therefore
increase lipid droplet formation62.
Another enzyme intimately involved
in lipid metabolism is hydroxyacyl-
CoA dehydrogenase alpha subunit
(HADHA). The role of HADHA in regulating
lipid droplet formation has been
examined in several models of ccRCC,
including the ccRCC cell line 786-O. In
this cell line, OmicsNet and STRING
analysis revealed an abundance of enzymes
involved in lipid metabolism,
including HADHA and acetyl-CoA
acetyltransferase 2 (ACAT2), exist in a
network. Additionally, several direct
protein-protein interactions were identified
in this network, including a link
between HADHA and ACAT2, which
allows them to interact with substrates
in a coordinated manner69, 70. HADHA
was shown to activate ACAT2, an enzyme
directly involved in lipid breakdown,
so at low HADHA levels, there
are low levels of lipid breakdown causing
lipid stores to be maintained, which
is associated with ccRCC tumor cell
proliferation69. In a separate study, it
was confirmed that there is downregulation
of both HADHA and ACAT2
in ccRCC patient tissues and that this
downregulation of HADHA expression
in ccRCC tumors was associated with
better patient survival70. The goal in
studying lipid metabolism of ccRCC is
to identify opportunities to intervene
therapeutically inhibiting the rapid proliferation
and expansion of cells present
in the tumor, as well as impeding formation
of new cells. KLF6, PLIN2, HIF-
2α, HADHA, ACAT, and CPT1A are
only a few of the lipid regulators that
have been identified for discussion in
this review, but the findings linked to
these mediators suggest avenues that
effect lipid droplet buildup could be
attractive targets for metabolic factors
incorporated into ccRCC prognosis and
treatment.
Cholesterol Metabolism
The clear cell phenotype is characterized
by lipid buildup, but recent studies
have shown that high-density lipoprotein
(HDL) cholesterol is accumulated
in the highest levels within ccRCC tissues.
HDL-cholesterol is also seen in
higher amount in ccRCC tumoral cells
compared the surrounding non-malignant
kidney tissues71–73. The deregulation
of cholesterol compounds with the
accumulation of other lipids to stabilize
the membrane of the tumoral cells and
increases tumorigenesis when it cannot
be regulated properly. In multiple
studies, cholesterol synthesis did not
appear to be affected, which suggests
that the cholesterol buildup seen within
the cells is due to exogenous cholesterol
influx and endogenous cholesterol
efflux71, 74. Cholesterol was also discovered
to play a role in promoting metastasis
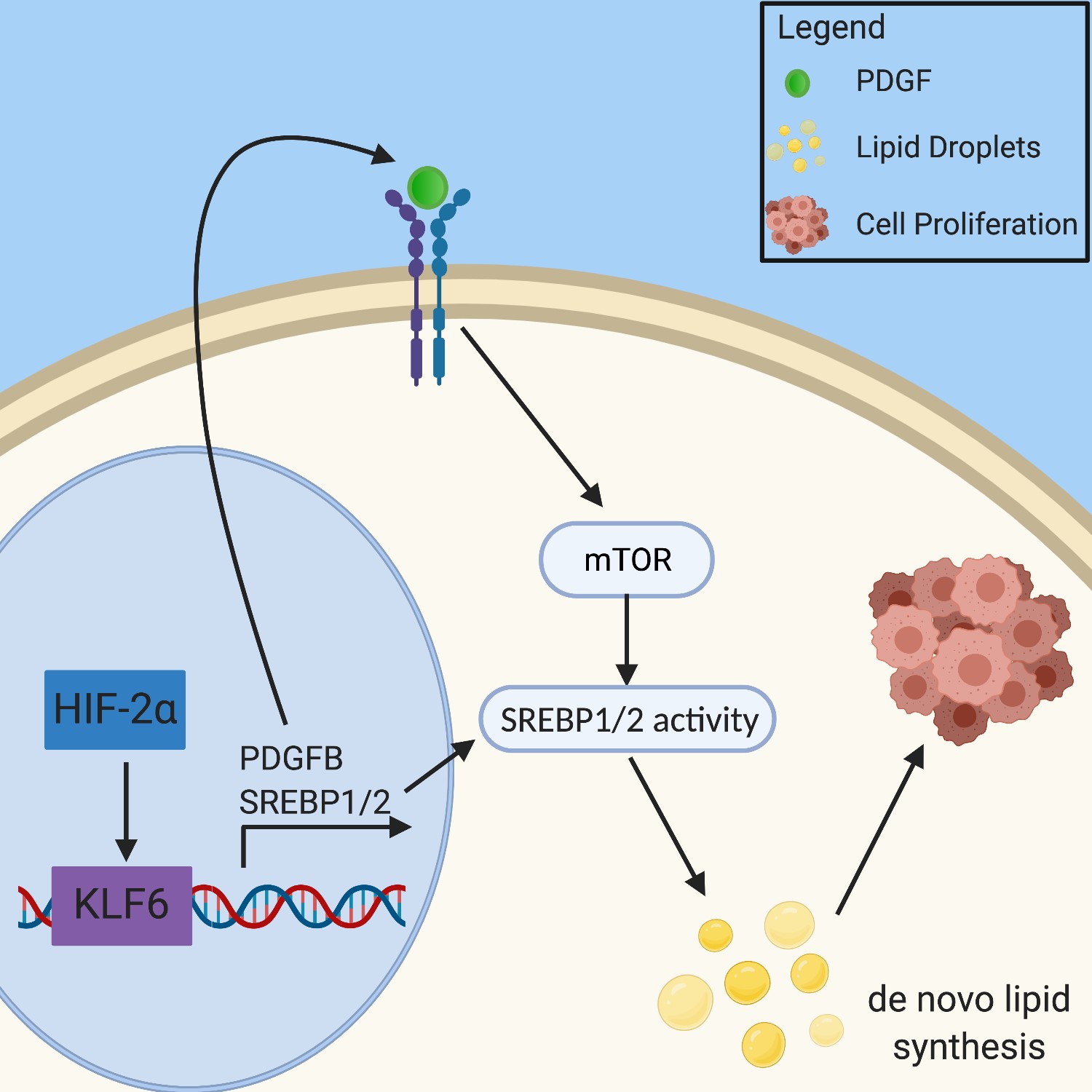
of ccRCC75. Hypoxia effects fatty
acid saturation via the oxygen dependent
enzyme stearoyl-CoA desaturase
(SCD). SCD under hypoxic conditions
is inhibited, which leads to a buildup of
fatty acid precursors in the cell76. This
leads to disruption of the endoplasmic
reticulum and induces apoptosis76–78
(Figure 3).
A recent study demonstrated
how cholesterol buildup in tumoral
cells is due to the uptake of cholesterol
rather than synthesis71. The cholesterol
synthesis rate limiting enzyme HMGCR
was inhibited in tumors containing
higher levels of cholesterol, suggesting
that cholesterol de novo synthesis is unlikely
to be occurring in the tumor cell.
Furthermore, they showed that the receptor
for HDL-cholesterol, scavenger
receptor B1 (SR-B1), which is usually in
very low concentrations in the cell, had
elevated levels in tumors containing
high levels of cholesterol71.
Another study explored the
difference in predicted treatment efficacy
by targeting the transcription factor
receptor, liver X receptor (LXR) with an
agonist versus an inverse agonist. The
agonist used was LXR623 and the inverse
agonist was SR9243. Both inhibited
cell proliferation and induced apoptosis,
but by different mechanisms.
LXR623 killed tumor cells by promoting
cholesterol efflux and inhibiting
cholesterol influx. SR9243 upregulated
the HMOX2 gene which reduced the
angiogenic potential and proliferation,
and it also caused a decrease in intracellular
triglycerides. Neither affected the
cholesterol synthesis pathway74. This
makes these therapeutic targets attractive
for future consideration because
the synthesis of cholesterol is the main
mechanism of cholesterol accumulation
in normal cells. Since there is little to no
new synthesis of cholesterol in ccRCC
tumoral cells, but rather change in how
much cholesterol is moving into the cell,
the cholesterol receptors can be targets
for therapeutic intervention with a potential
window of specificity for tumor
cells in this case.
Although high cholesterol levels
are common to all ccRCC tumors,
cholesterol levels in the body have also
been associated with outcome in the
case of ccRCC. High HDL-cholesterol
levels were correlated with better outcomes
and can act as a similar predictor
in other forms of cancer as well79. The
mechanism by which this is achieved
is believed to be that the higher HDLcholesterol
in the body, the less uptake
of low-density lipoproteins (LDL) by tumor
cells which would suggest that there
is less lipid support for tumor growth75,
although additional work is needed to
understand this association more fully.
Statins, which are clinically used to lower
LDL levels in patients, have been considered
as a possible therapeutic target.
A recent study showed that treatment
with statins in VHL-deficient ccRCC
elicited promising early findings and
suggested that the observed lethality is
HIF dependent, highlighting statins as
promising therapeutic tools80.
Future Directions
Further analysis is needed for current
treatments that can augment the current
armamentarium for ccRCC. An
area for growth in the research of therapeutic
treatments is in targeting the
metabolic dependencies, such as glycolysis,
lipid, and cholesterol metabolism
pathways, that discriminate ccRCC
cells from normal tissues, or that reveal
cellular adaptations associated with
disease progression.
In order to control glycogen
metabolism in a favorable manner,
promoting glycogen breakdown while
simultaneously preventing glucose metabolism
and glycogen synthesis is the
goal. Glucagon is a natural substance in
the body that accomplishes this by activating
glycogen phosphorylase through
the activity of protein kinase A. Finding
a molecular target that can mimic this
pathway specifically in ccRCC could be
a direction worth pursuing. It is worth
noting, glycogen breakdown to glucose-
1-phosphate feeds into glycolysis
which could fuel growth, so another
approach could involve a combination
of nutrient restriction and current
frontline therapies that impede cell
growth and metabolism. There are no
current studies that have examined the
effects of dietary restrictions on ccRCC
patients, but a correlation between BMI
and the presence or absence of a VHL
mutation in ccRCC patients has been
observed81.
In considering lipid and cholesterol
metabolism for therapeutic development,
it is known how the inhibition
of SCD leads to cholesterol accumulation,
but there have been no further
studies completed to show the relationship
between VHL mutations and
cholesterol synthesis. Secondly, while
statins look to be a promising target
and have shown to inhibit the proliferation
of VHL-deficient ccRCC in vitro
and in vivo, further analysis needs to
be done on the efficacy, mode of action,
and safety of these treatments. Also,
since dietary lipid intake was shown to
effect lipid buildup in the kidneys, further
investigation should be conducted
to determine outcomes when cholesterol
treatments are compounded with dietary
and host factors.
There is minimal literature
in ccRCC investigating the role of acetate
metabolism, an important branch
of acetyl-CoA production and a key
contributor to lipogenesis. Therefore,
acetate metabolism and the enzyme
acetate-dependent acetyl-CoA synthetase
2 (ACSS2) could be a potential
therapeutic target. While this has not
been explored in ccRCC, researchers
have demonstrated in other tissues that
inhibition of ACSS2 leads to the inhibition
of lipid metabolism, changes to
histone acetylation, and reduced tumor
growth82. ACSS2 is required for acetate
uptake and ACSS2 deficient mice were
shown to have decreased liver tumor
formation83. Nuclear ACSS2 synthesizes
acetyl-CoA for histone acetylation,
which activates lysozyme biogenesis84
Interestingly, it has been shown that
acetyl-CoA derived from ACSS2 is required
for the acetylation of HIF-2α and
results in optimal signaling85. These
factors make ACSS2 an enzyme of interest
for further investigation.
In summary, bioenergetic metabolism
has long been recognized as
a differentiating feature of ccRCC, and
as we gain insights into these pathways
and methods to intervene. Future work
to incorporate these strategies in combination
or in sequence with existing
therapies will be a major opportunity
to impact this metabolically driven
disease.
1. E. van den Berg, “Renal Cell Carcinoma,” in
Brenner’s Encyclopedia of Genetics (Second 27 Edition),
S. Maloy and K. Hughes, Eds. San Diego: Academic
Press, 2013, pp. 130–132. 28
2. M. B. Atkins and N. M. Tannir, “Current
and emerging therapies for first-line treatment of 29
metastatic clear cell renal cell carcinoma,” Cancer
Treatment Reviews, vol. 70, pp. 127– 137, Nov. 2018. 31
3. X. Yao et al., “Two Distinct Types of Blood Vessels
in Clear Cell Renal Cell Carcinoma Have 32 Contrasting
Prognostic Implications,” American Association for
Cancer Research, vol. 13, 33 no. 1, pp. 161–169, Jan. 2007.
34
4. W. Xu, M. B. Atkins, and D. F. McDermott,
“Checkpoint inhibitor immunotherapy in kidney
cancer,” Nature Reviews Urology, vol. 17, no. 3, Art. no.
3, Mar. 2020, doi: 36 10.1038/s41585-020-0282-3. 37
5. C. J. Creighton et al., “Comprehensive molecular
characterization of clear cell renal cell 38 carcinoma,”
Nature, vol. 499, no. 7456, Art. no. 7456, Jul. 2013, doi: 39
10.1038/nature12222.
6. W. M. Linehan et al., “The Metabolic Basis of
Kidney Cancer,” Cancer Discov, vol. 9, no. 8, 41 pp. 1006–
1021, Aug. 2019, doi: 10.1158/2159-8290.CD-18-1354.
7. C. J. Ricketts et al., “The Cancer Genome Atlas
Comprehensive Molecular Characterization of Renal Cell
Carcinoma,” Cell Reports, vol. 23, no. 1, pp. 313-326.e5,
Apr. 2018, doi: 10.1016/j.celrep.2018.03.075.
8. H. I. Wettersten et al., “Grade-Dependent
Metabolic Reprogramming in Kidney Cancer Revealed
by Combined Proteomics and Metabolomics Analysis,”
Cancer Res, vol. 75, no. 12, pp. 2541–2552, Jun. 2015, doi:
10.1158/0008-5472.CAN-14-1703.
9. A. A. Hakimi et al., “An Integrated Metabolic
Atlas of Clear Cell Renal Cell Carcinoma,” Cancer Cell,
vol. 29, no. 1, pp. 104–116, Jan. 2016, doi: 10.1016/j.
ccell.2015.12.004.
10. K. D. Courtney et al., “Isotope Tracing of
Human Clear Cell Renal Cell Carcinomas Demonstrates
Suppressed Glucose Oxidation In Vivo,” Cell Metabolism,
vol. 28, no. 5, pp. 793-800.e2, Nov. 2018, doi: 10.1016/j.
cmet.2018.07.020.
11. A. R. Mullen et al., “Reductive carboxylation
supports growth in tumor cells with defective
mitochondria,” Nature, vol. 481, no. 7381, pp. 385–388,
Nov. 2011, doi: 10.1038/nature10642.
12. C. M. Metallo et al., “Reductive glutamine
metabolism by IDH1 mediates lipogenesis under
hypoxia,” Nature, vol. 481, no. 7381, pp. 380–384, Nov.
2011, doi: 10.1038/nature10602.
13. P. A. Gameiro et al., “In Vivo HIF-Mediated
Reductive Carboxylation Is Regulated by Citrate Levels
and Sensitizes VHL-Deficient Cells to Glutamine
Deprivation,” Cell Metab, vol. 17, no. 3, pp. 372–385, Mar.
2013, doi: 10.1016/j.cmet.2013.02.002.
14. C. L. Cowey and W. K. Rathmell, “VHL gene
mutations in renal cell carcinoma: role as a biomarker
of disease outcome and drug efficacy,” Curr Oncol
Rep, vol. 11, no. 2, pp. 94– 101, Mar. 2009, doi: 10.1007/
s11912-009-0015-5.
15. W. K. Rathmell and S. Chen, “VHL
inactivation in renal cell carcinoma: implications for
diagnosis, prognosis and treatment,” Expert Rev
Anticancer Ther, vol. 8, no. 1, pp. 63–73, Jan. 2008, doi:
10.1586/14737140.8.1.63.
16. C. Shen and W. G. Kaelin, “The VHL/HIF
axis in clear cell renal carcinoma,” Seminars in Cancer
Biology, vol. 23, no. 1, pp. 18–25, Feb. 2013, doi: 10.1016/j.
semcancer.2012.06.001.
17. J. C. Chappell, L. B. Payne, and W. K. Rathmell,
“Hypoxia, angiogenesis, and metabolism in the
hereditary kidney cancers,” J Clin Invest, vol. 129, no. 2,
pp. 442–451, doi: 10.1172/JCI120855.
18. Z. A. Bacigalupa and W. K. Rathmell, “Beyond
glycolysis: Hypoxia signaling as a master regulator of
alternative metabolic pathways and the implications in
clear cell renal cell carcinoma,” Cancer Letters, vol. 489,
pp. 19–28, Oct. 2020, doi: 10.1016/j.canlet.2020.05.034.
19. L. E. Moore et al., “Von Hippel-Lindau (VHL)
Inactivation in Sporadic Clear Cell Renal Cancer:
Associations with Germline VHL Polymorphisms and
Etiologic Risk Factors,” PLOS Genetics, vol. 7, no. 10, p.
e1002312, Oct. 2011, doi: 10.1371/journal.pgen.1002312.
20. C.-N. Qian, D. Huang, B. Wondergem, and B.
T. Teh, “Complexity of tumor vasculature in clear cell
renal cell carcinoma,” American Cancer Society, vol.
115, no. S10, Apr. 2009,Online. Available: https://doi.
org/10.1002/cncr.24238.
21. J. D. Weyandt, C. B. Thompson, A. J. Giaccia,
and W. K. Rathmell, “Metabolic Alterations In Cancer
and Their Potential As Therapeutic Targets,” Am Soc
Clin Oncol Educ Book, vol. 37, pp. 825–832, 2017, doi:
10.14694/EDBK_175561.
22. G. L. Semenza, “HIF-1 mediates the Warburg
effect in clear cell renal carcinoma,” J Bioenerg Biomembr,
vol. 39, no. 3, pp. 231–234, Jun. 2007, doi: 10.1007/
s10863-0079081-
2.
23. J. D. Gordan et al., “HIF-a effects on c-Myc
distinguish two subtypes of sporadic VHL-deficient clear
cell renal carcinoma,” Cancer Cell, vol. 14, no. 6, pp. 435–
446, Dec. 2008, doi: 10.1016/j.ccr.2008.10.016.
24. B. Shuch, W. M. Linehan, and R. Srinivasan,
“Aerobic glycolysis: a novel target in kidney cancer,”
Expert Review of Anticancer Therapy, vol. 13, no. 6, pp.
711–719, Jun. 2013, doi: 10.1586/era.13.57.
25. C.-J. Hu, L.-Y. Wang, L. A. Chodosh, B. Keith,
and M. C. Simon, “Differential Roles of Hypoxia-
Inducible Factor 1a (HIF-1a) and HIF-2a in Hypoxic
Gene Regulation,” Molecular and Cellular Biology,
vol. 23, no. 24, pp. 9361–9374, Dec. 2003, doi: 10.1128/
MCB.23.24.9361-9374.2003.
26. T. K. Choueiri and W. G. Kaelin, “Targeting
the HIF2–VEGF axis in renal cell carcinoma,” Nat Med,
vol. 26, no. 10, pp. 1519–1530, Oct. 2020, doi: 10.1038/
s41591-020-1093-z.
27. R. Srinivasan, C. J. Ricketts, C. Sourbier, and W.
M. Linehan, “New Strategies in Renal Cell Carcinoma:
Targeting the Genetic and Metabolic Basis of Disease,”
Clin Cancer Res, vol. 21, no. 1, pp. 10–17, Jan. 2015, doi:
10.1158/1078-0432.CCR-13-2993.
28. K. Kondo, W. Y. Kim, M. Lechpammer, and W.
G. Kaelin, “Inhibition of HIF2a Is Sufficient to Suppress
pVHL-Defective Tumor Growth,” PLoS Biol, vol. 1, no. 3,
Dec. 2003, doi: 10.1371/journal.pbio.0000083.
29. M. Zimmer, D. Doucette, N. Siddiqui, and O.
Iliopoulos, “Inhibition of Hypoxia-Inducible Factor Is
Sufficient for Growth Suppression of VHL-/- Tumors1
1 NIH grant R29CA7835806
(O. I.), Bertucci Fund for
Urologic Malignancies (O. I.), David P. Foss Fund (O.
I.), and VHL Family Alliance 2003 award (M. Z.).,” Mol
Cancer Res, vol. 2, no. 2, pp. 89–95, Feb. 2004.
30. M. Zimmer et al., “Small molecule inhibitors
of HIF-2a translation link its 5’-UTR Iron-Responsive
Element (IRE) to oxygen sensing,” Mol Cell, vol. 32, no. 6,
pp. 838–848, Dec. 2008, doi: 10.1016/j.molcel.2008.12.004.
31. M. Zimmer et al., “The Connectivity Map links
Iron Response Protein-1 (IRP1)-mediated inhibition of
HIF2a translation to the anti-inflammatory 15-deoxy-
.12,14-Prostaglandin J2,” Cancer Res, vol. 70, no. 8,
pp. 3071–3079, Apr. 2010, doi: 10.1158/0008-5472.
CAN09-
2877.
32. C. Sourbier et al., “Targeting HIF2a Translation
with Tempol in VHL-Deficient Clear Cell Renal Cell
Carcinoma,” Oncotarget, vol. 3, no. 11, pp. 1472–1482,
Nov. 2012.
33. M. Sanchez, B. Galy, M. U. Muckenthaler, and
M. W. Hentze, “Iron-regulatory proteins limit hypoxiainducible
factor-2alpha expression in iron deficiency,”
Nat Struct Mol Biol, vol. 14, no. 5, pp. 420–426, May 2007,
doi: 10.1038/nsmb1222.
34. K. D. Courtney et al., “HIF-2 Complex
Dissociation, Target Inhibition, and Acquired Resistance
with PT2385, a First-in-Class HIF-2 Inhibitor in Clear Cell
Renal Cell Carcinoma Patients,” Clin Cancer Res, vol.
26, no. 4, pp. 793–803, Feb. 2020, doi: 10.1158/10780432.
CCR-19-1459.
35. R. Xu et al., “3-(1S,2S,3R)-2,3-Difluoro-
1-hydroxy-7-methylsulfonylindan-4-yloxy-5fluorobenzonitrile
(PT2977), a Hypoxia-Inducible Factor
2a (HIF-2a) Inhibitor for the Treatment of Clear Cell
Renal Cell Carcinoma,” J Med Chem, vol. 62, no. 15, pp.
6876– 6893, 08 2019, doi: 10.1021/acs.jmedchem.9b00719.
36. K. D. Courtney et al., “Phase I Dose-Escalation
Trial of PT2385, a First-in-Class Hypoxia-Inducible
Factor-2a Antagonist in Patients With Previously Treated
Advanced Clear Cell Renal Cell Carcinoma,” J Clin
Oncol, vol. 36, no. 9, pp. 867–874, Mar. 2018, doi: 10.1200/
JCO.2017.74.2627.
37. G. Yang et al., “Glucuronidation: Driving
Factors and Their Impact on Glucuronide Disposition,”
Drug Metab Rev, vol. 49, no. 2, pp. 105–138, May 2017,doi: 10.1080/03602532.2017.1293682.
38. K. P. Papadopoulos, E. Jonasch, N. J. Zojwalla,
K. Wang, and T. M. Bauer, “A first-in-human phase 1
dose-escalation trial of the oral HIF-2a inhibitor PT2977
in patients with advanced solid tumors.,” JCO, vol. 36,
no. 15_suppl, pp. 2508–2508, May 2018, doi: 10.1200/
JCO.2018.36.15_suppl.2508.
39. T. K. Choueiri et al., “Phase I/II study of
the oral HIF-2 a inhibitor MK-6482 in patients with
advanced clear cell renal cell carcinoma (RCC).,” JCO,
vol. 38, no. 6_suppl, pp. 611– 611, Feb. 2020, doi: 10.1200/
JCO.2020.38.6_suppl.611.
40. E. Jonasch et al., “Phase II study of the oral
HIF-2a inhibitor MK-6482 for Von Hippel-Lindau
disease–associated renal cell carcinoma.,” JCO, vol. 38,
no. 15_suppl, pp. 5003– 5003, May 2020, doi: 10.1200/
JCO.2020.38.15_suppl.5003.
41. T. K. Choueiri et al., “Phase III study of the
hypoxia-inducible factor 2a (HIF-2a) inhibitor MK-6482
versus everolimus in previously treated patients with
advanced clear cell renal cell carcinoma (ccRCC).,” JCO,
vol. 38, no. 15_suppl, pp. TPS5094–TPS5094, May 2020,
doi: 10.1200/JCO.2020.38.15_suppl.TPS5094.
42. W. Chen et al., “Targeting Renal Cell Carcinoma
with a HIF-2 antagonist,” Nature, vol. 539, no. 7627, pp.
112–117, Nov. 2016, doi: 10.1038/nature19796.
43. L. Zhou et al., “Targeting MET and AXL
overcomes resistance to sunitinib therapy in renal cell
carcinoma,” Oncogene, vol. 35, no. 21, pp. 2687–2697,
May 2016, doi: 10.1038/onc.2015.343.
44. E. B. Rankin et al., “Direct regulation of GAS6/
AXL signaling by HIF promotes renal metastasis
through SRC and MET,” Proc Natl Acad Sci U S A, vol.
111, no. 37, pp. 13373– 13378, Sep. 2014, doi: 10.1073/
pnas.1404848111.
45. F. Shojaei et al., “HGF/c-Met Acts as an
Alternative Angiogenic Pathway in Sunitinib-Resistant
Tumors,” Cancer Res, vol. 70, no. 24, pp. 10090–10100,
Dec. 2010, doi: 10.1158/0008-5472.CAN-10-0489.
46. B. Sennino et al., “Suppression of tumor
invasion and metastasis by concurrent inhibition of
c-Met and VEGF signaling in pancreatic neuroendocrine
tumors,” Cancer Discov, vol. 2, no. 3, pp. 270–287, Mar.
2012, doi: 10.1158/2159-8290.CD-11-0240.
47. E. Ciamporcero et al., “Combination strategy
targeting VEGF and HGF/c-met in human renal cell
carcinoma models,” Mol Cancer Ther, vol. 14, no. 1, pp.
101–110, Jan. 2015, doi: 10.1158/1535-7163.MCT-14-0094.
48. M. Laplante and D. M. Sabatini, “mTOR
signaling in growth control and disease,” Cell, vol.
149, no. 2, pp. 274–293, Apr. 2012, doi: 10.1016/j.
cell.2012.03.017.
49. E. J. Brown et al., “A mammalian protein
targeted by G1-arresting rapamycin-receptor complex,”
Nature, vol. 369, no. 6483, pp. 756–758, Jun. 1994, doi:
10.1038/369756a0.
50. D. M. Sabatini, H. Erdjument-Bromage, M. Lui, P.
Tempst, and S. H. Snyder, “RAFT1: a mammalian protein
that binds to FKBP12 in a rapamycin-dependent fashion
and is homologous to yeast TORs,” Cell, vol. 78, no. 1, pp.
35–43, Jul. 1994, doi: 10.1016/00928674(
94)90570-3.
51. J. Li, S. G. Kim, and J. Blenis, “Rapamycin: one
drug, many effects,” Cell Metab, vol. 19, no. 3, pp. 373–
379, Mar. 2014, doi: 10.1016/j.cmet.2014.01.001.
52. Y. Lai et al., “The tumour microenvironment
and metabolism in renal cell carcinoma targeted or
immune therapy,” Journal of Cellular Physiology, vol.
n/a, no. n/a, doi: 10.1002/jcp.29969.
53. Y. Lu et al., “The different expression of glycogen
phosphorylases in renal clear cell renal carcinoma and
chromophobe renal carcinoma,” Clin Proteomics, vol. 17,
Feb. 2020, doi: 10.1186/s12014-020-9270-0.
54. C. Bianchi et al., “The glucose and lipid
metabolism reprogramming is grade-dependent in
clear cell renal cell carcinoma primary cultures and is
targetable to modulate cell viability and proliferation,”
Oncotarget, vol. 8, no. 69, pp. 113502–113515, Dec. 2017,
doi: 10.18632/oncotarget.23056.
55. A. R. Brannon et al., “Molecular Stratification
of Clear Cell Renal Cell Carcinoma by Consensus
Clustering Reveals Distinct Subtypes and Survival
Patterns,” Genes Cancer, vol. 1, no. 2, pp. 152–163, Feb.
2010, doi: 10.1177/1947601909359929.
56. S. A. Brooks et al., “ClearCode34: A prognostic
risk predictor for localized clear cell renal cell carcinoma,”
Eur Urol, vol. 66, no. 1, pp. 77–84, Jul. 2014, doi: 10.1016/j.
eururo.2014.02.035.
57. S. Ribback et al., “PI3K/AKT/mTOR pathway
plays a major pathogenetic role in glycogen accumulation
and tumor development in renal distal tubules of rats
and men,” Oncotarget, vol. 6, no. 15, pp. 13036–13048,
Apr. 2015.
58. Y. Lai et al., “Crosstalk between VEGFR and
other receptor tyrosine kinases for TKI therapy of
metastatic renal cell carcinoma,” Cancer Cell Int, vol. 18,
Mar. 2018, doi: 10.1186/s12935-018-0530-2.
59. S. Chen et al., “GYS1 induces glycogen
accumulation and promotes tumor progression via
the NF-.B pathway in Clear Cell Renal Carcinoma,”
Theranostics, vol. 10, no. 20, pp. 9186–9199, Jul. 2020, doi:
10.7150/thno.46825.
60. B. Li et al., “Fructose-1, 6-bisphosphatase
opposes renal carcinoma progression,” Nature, vol.
513, no. 7517, pp. 251–255, Sep. 2014, doi: 10.1038/
nature13557.
61. J. C. van der Mijn et al., “Combined
Metabolomics and Genome-Wide Transcriptomics
Analyses Show Multiple HIF1a-Induced Changes in
Lipid Metabolism in Early Stage Clear Cell Renal Cell
Carcinoma,” Transl Oncol, vol. 13, no. 2, pp. 177–185,
Dec. 2019, doi: 10.1016/j.tranon.2019.10.015.
62 W. Du et al., “HIF drives lipid deposition and
cancer in ccRCC via repression of fatty acid metabolism,”
Nat Commun, vol. 8, Nov. 2017, doi: 10.1038/
s41467-017-01965-8.
63. M. Bao, R. Shi, K. Zhang, Y. Zhao, Y. Wang,
and X. Bao, “Development of a membrane lipid
metabolism–based signature to predict overall survival
for personalized medicine in ccRCC patients,” EPMA
Journal, vol. 10, no. 4, pp. 383–393, Dec. 2019, doi:
10.1007/s13167-019-00189-8.
64. S. E. Syafruddin et al., “A KLF6-driven
transcriptional network links lipid homeostasis and
tumour growth in renal carcinoma,” Nat Commun, vol.
10, Mar. 2019, doi: 10.1038/s41467-019-09116-x.
65. A. Brauweiler et al., “RING-dependent tumor
suppression and G2/M arrest induced by the TRC8
hereditary kidney cancer gene,” Oncogene, vol. 26, no.
16, Art. no. 16, Apr. 2007, doi: 10.1038/sj.onc.1210017.
66. J. D. Horton et al., “Combined analysis of
oligonucleotide microarray data from transgenic and
knockout mice identifies direct SREBP target genes,”
PNAS, vol. 100, no. 21, pp. 12027–12032, Oct. 2003, doi:
10.1073/pnas.1534923100.
67. J. L. Goldstein, R. A. DeBose-Boyd, and M.
S. Brown, “Protein Sensors for Membrane Sterols,”
Cell, vol. 124, no. 1, pp. 35–46, Jan. 2006, doi: 10.1016/j.
cell.2005.12.022.
68. B. Qiu et al., “HIF-2a dependent lipid storage
promotes endoplasmic reticulum homeostasis in clear
cell renal cell carcinoma,” Cancer Discov, vol. 5, no. 6, pp.
652–667, Jun. 2015, doi: 10.1158/2159-8290.CD-14-1507.
69. S. Liu et al., “HADHA overexpression disrupts
lipid metabolism and inhibits tumor growth in clear
cell renal cell carcinoma,” Experimental Cell Research,
vol. 384, no. 1, p. 111558, Nov. 2019, doi: 10.1016/j.
yexcr.2019.111558.
70. Z. Zhao, J. Lu, L. Han, X. Wang, Q. Man, and
S. Liu, “Prognostic significance of two lipid metabolism
enzymes, HADHA and ACAT2, in clear cell renal cell
carcinoma,” Tumor Biol., vol. 37, no. 6, pp. 8121–8130,
Jun. 2016, doi: 10.1007/s13277-015-4720-4.
71. J. Kim, B. Thompson, S. Han, Y. Lotan, J. G.
McDonald, and J. Ye, “Uptake of HDL-cholesterol
contributes to lipid accumulation in clear cell renal
cell carcinoma,” Biochimica et Biophysica Acta (BBA)
-Molecular and Cell Biology of Lipids, vol. 1864, no. 12,
p. 158525, Dec. 2019, doi: 10.1016/j.bbalip.2019.158525.
72. Y. Zhang et al., “Addressing metabolic
heterogeneity in clear cell renal cell carcinoma with
quantitative Dixon MRI,” JCI Insight, vol. 2, no. 15, doi:
10.1172/jci.insight.94278.
73. K. Saito et al., “Lipidomic Signatures and
Associated Transcriptomic Profiles of Clear Cell Renal
Cell Carcinoma,” Scientific Reports, vol. 6, no. 1, Art. no.
1, Jun. 2016, doi: 10.1038/srep28932.
74. G. Wu et al., “Targeting the transcription factor
receptor LXR to treat clear cell renal cell carcinoma:
agonist or inverse agonist?,” Cell Death Dis, vol. 10, no.
6, May 2019, doi: 10.1038/s41419-019-1654-6.
75. H. Yang et al., “Exploring the mechanism of
clear cell renal cell carcinoma metastasis and key genes
based on multi-tool joint analysis,” Gene, vol. 720, p.
144103, Dec. 2019, doi: 10.1016/j.gene.2019.144103.
76. D. Ackerman et al., “Triglycerides Promote
Lipid Homeostasis during Hypoxic Stress by Balancing
Fatty Acid Saturation,” Cell Rep, vol. 24, no. 10, pp. 2596-
2605.e5, Sep. 2018, doi: 10.1016/j.celrep.2018.08.015.
77. J. J. Kamphorst et al., “Hypoxic and Rastransformed
cells support growth by scavenging
unsaturated fatty acids from lysophospholipids,” Proc
Natl Acad Sci U S A, vol. 110, no. 22, pp. 8882–8887, May
2013, doi: 10.1073/pnas.1307237110.
78. R. M. Young et al., “Dysregulated mTORC1
renders cells critically dependent on desaturated lipids
for survival under tumor-like stress,” Genes Dev,
vol. 27, no. 10, pp. 1115–1131, May 2013, doi: 10.1101/
gad.198630.112.
79. B. Hao, X. Peng, B. Bi, M. Yu, C. Sang, and Z.
Chen, “Preoperative serum high-density lipoprotein
cholesterol as a predictor of poor survival in patients
with clear cell renal cell cancer:,” The International
Journal of Biological Markers, Mar. 2019, doi:
10.1177/1724600819831404.
80. J. M. Thompson et al., “Targeting the mevalonate
pathway suppresses VHL-deficient CCRCC
through a
HIF-dependent mechanism,” Mol Cancer Ther, vol. 17,
no. 8, pp. 1781– 1792, Aug. 2018, doi: 10.1158/1535-7163.
MCT-17-1076.
81. K. M. Smits et al., “Body Mass Index and von
Hippel-Lindau Gene Mutations in Clear-cell Renal
Cancer: Results of the Netherlands Cohort Study on Diet
and Cancer,” Annals of Epidemiology, vol. 20, no. 5, pp.
401–404, May 2010, doi: 10.1016/j.annepidem.2010.01.010.
82. Y. Ni et al., “miR-15a-5p inhibits metastasis and
lipid metabolism by suppressing histone acetylation in
lung cancer,” Free Radic Biol Med, vol. 161, pp. 150–162,
Oct. 2020, doi: 10.1016/j.freeradbiomed.2020.10.009.
83. S. A. Comerford et al., “Acetate Dependence of
Tumors,” Cell, vol. 159, no. 7, pp. 1591– 1602, Dec. 2014,
doi: 10.1016/j.cell.2014.11.020.
84. X. Li, X. Qian, and Z. Lu, “Local histone
acetylation by ACSS2 promotes gene transcription for
lysosomal biogenesis and autophagy,” Autophagy,
vol. 13, no. 10, pp. 1790–1791, Aug. 2017, doi:
10.1080/15548627.2017.1349581.
85. J. S. Nagati, M. Xu, T. Garcia, S. A. Comerford,
R. E. Hammer, and J. A. Garcia, “A substitution mutation
in a conserved domain of mammalian acetate-dependent
acetyl CoA synthetase 2 results in destabilized protein
and impaired HIF-2 signaling,” PLoS One, vol. 14, no.
11, p. e0225105, 2019, doi: 10.1371/journal.pone.0225105.
Correspondence: W. Kimryn Rathmell, M.D., Ph.D., Vanderbilt University Medical Center
Department of Medicine, 1161 21st Avenue South Suite D-3100, Medical Center North,
Nashville, TN 37232. Phone: 615-343-8701; Fax: 616-343-2551; Email: Kimryn.Rathmell@
VUMC.org